Input-file format¶
The input file consists of five parameter blocks named [model], [system], [impurity_solver], [control], and [tool].
The following table shows which blocks are used by each program.
|
|
|
|
|
|---|---|---|---|---|
[model] |
Yes |
Yes |
Yes |
Yes |
[system] |
Yes |
Yes |
Yes |
|
[impurity_solver] |
Yes |
Yes |
||
[control] |
Yes |
|||
[tool] |
Yes |
Yes |
||
[mpi] |
Yes |
Yes |
For example, we can see that dcore_pre needs to be re-executed only when [model] block is changed.
The parameters included in each block are explained below.
[model] block¶
This block includes parameters for defining a model to be solved.
Name |
Type |
Default |
Description |
|---|---|---|---|
seedname |
String |
dcore |
Name of the system. The model HDF5 file will be seedname.h5. |
lattice |
String |
chain |
Chosen from “chain”, “square”, “cubic”, “bethe”, “wannier90”, and “external” |
t |
Float |
1.0 |
Transfer integral (Nearest neighbor) |
t’ |
Float |
0.0 |
Transfer integral (Second nearest) |
nelec |
Float |
1.0 |
Number of electrons per unit cell. |
norb |
String |
1 |
Number of orbitals at each correlated shell (ncor integers separated by commas or spaces.) |
ncor |
Integer |
1 |
Number of correlated shells in a unit cell (for lattice = wannier90). |
corr_to_inequiv |
String |
None |
Mapping from correlated shells to equivalent shells (for lattice = wannier90) |
bvec |
String |
[(1.0,0.0,0.0),(0.0,1.0,0.0),(0.0,0.0,1.0)] |
Reciprocal lattice vectors in arbitrary unit. |
nk |
Integer |
8 |
Number of k along each line |
nk0 |
Integer |
0 |
Number of k along b_0 (for lattice = wannier90, external) |
nk1 |
Integer |
0 |
Number of k along b_1 (for lattice = wannier90, external) |
nk2 |
Integer |
0 |
Number of k along b_2 (for lattice = wannier90, external) |
spin_orbit |
Bool |
False |
Whether the spin-orbit case. |
interaction |
String |
kanamori |
Chosen from “slater_uj”, “slater_f”, “kanamori”, “respack” (See below) |
density_density |
Bool |
False |
If true, only the density-density part of the interaction is used (See below). |
kanamori |
String |
None |
U (Diagonal Coulomb pot.), U’ (Off-diagonal Coulomb pot.) and J (Hund coupling) (See below). |
slater_f |
String |
None |
Angular momentum, Slater integrals F (See below). |
slater_uj |
String |
None |
Angular momentum, Slater integrals in U and J (See below). |
local_potential_matrix |
String |
None |
dict of {ish: ‘filename’} to specify local potential matrix of ish-th shell |
local_potential_factor |
String |
1.0 |
Prefactors to the local potential matrix (float or list with len=ncor) |
lattice¶
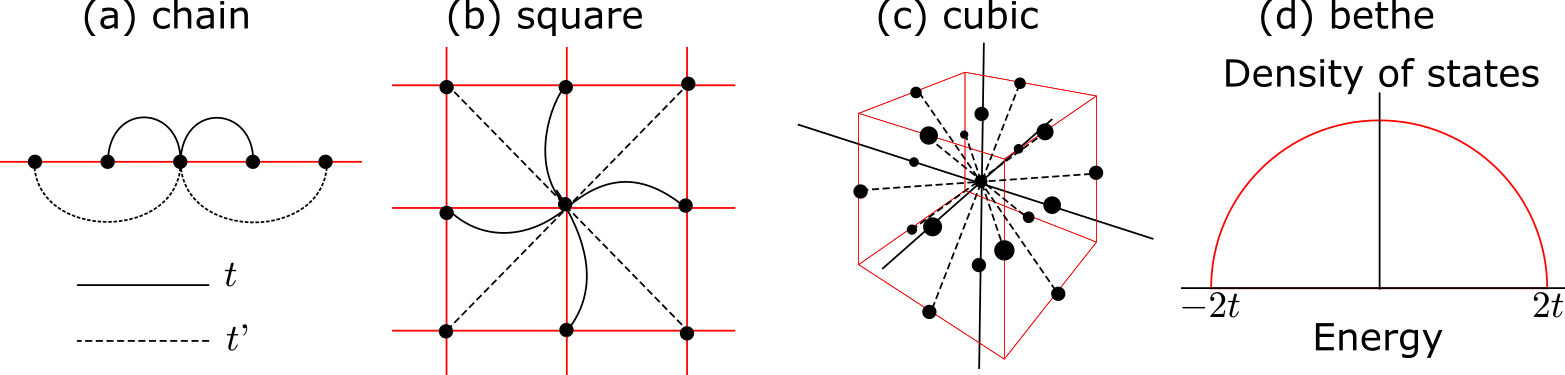
For model calculations, the following preset models are defined:
chain
square
cubic
bethe (semicircular DOS with energy ranges [-2t:2t])
For DFT+DMFT calculations, hopping parameters in the Wannier90 format can be imported by
wannier90
Place the Wannier90 file in the current directory with the name seedname_hr.dat.
For experts, the lattice data may be prepared by your own. In this case, use
external
In this mode, you should make all necessary data in
dft_inputgroup of seedname.h5. The data structure follows DFTTools. For details, see the reference manual of DFTTools.The pre-process
dcore_predoes not touch the data indft_inputgroup, and write only additional data such as interactions intoDCoregroup.
interaction¶
Model Hamiltonian is defined as
where
The interaction matrix \(U^{i}_{\alpha \beta \gamma \delta}\)
is specified by the parameter interaction.
If
interaction = kanamoriIn this case, the Kanamori-type interaction is used, i.e.
\[\begin{split}\begin{align} U_{\alpha \alpha \alpha \alpha} &= U, \\ U_{\alpha \beta \alpha \beta} &= U' \qquad (\alpha \neq \beta), \\ U_{\alpha \beta \beta \alpha} &= J \qquad (\alpha \neq \beta), \\ U_{\alpha \alpha \beta \beta} &= J \qquad (\alpha \neq \beta), \end{align}\end{split}\]where \(U, U', J\) at each correlated shell are specified by the parameter
kanamoriasinteraction = kanamori kanamori = [(U_1, U'_1, J_1), (U_2, U'_2, J_2), ... ]
For example, if there are two correlated shells that have \((U, U', J) = (4, 2, 1)\) and \((U, U', J) = (6, 3, 1.5)\), respectively, you need to set the input parameters as
interaction = kanamori kanamori = [(4.0, 2.0, 1.0), (6.0, 3.0, 1.5)]
If
interaction = slater_fIn this case, the interaction matrix is constructed by the effective Slater integrals \(F_0, F_2, F_4, F_6\). These Slater integrals and the angular momentum at each correlated shell are specified by the parameter
slater_fas followsinteraction = slater_f slater_f = [(angular_momentum, F_0, F_2, F_4, F_6), ... ]
For example, if there are two correlated shells, one has d-orbital with \((F_0, F_2, F_4) = (2, 1, 0.5)\) and the other has p-orbital with \((F_0, F_2) = (3, 1.5)\), you need to set the input parameter as
interaction = slater_f slater_f = [(2, 2.0, 1.0, 0.5, 0.0), (1, 3.0, 1.5 0.0, 0.0)]
Note
You must specify all of \(F_0, F_2, F_4, F_6\).
If
interaction = slater_ujIn this case, the Slater-type interaction is used. The effective Slater integrals are computed with the following formulae:
\(l = 1\)
\[F_0 = U, \quad F_2 = 5 J\]\(l=2\)
\[F_0 = U, \quad F_2 = \frac{14 J}{1.0 + 0.63},\quad F_4 = 0.63 F_2\]\(l=3\)
\[F_0 = U, \quad F_2 = \frac{6435 J}{286 + 195 \times 451 / 675 + 250 \times 1001 / 2025},\quad F_4 = \frac{451 F_2}{675},\quad F_6 = \frac{1001 F_2}{2025}\]
The \(U\), \(J\) and the angular momentum at each correlated shell are specified by the parameter
slater_ujasinteraction = slater_uj slater_uj = [(angular_momentum1, U1, J1), (angular_momentum2, U2, J2), ... ]
If
interaction = respackUse the output by RESPACK. Under construction.
If you want to treat only the density-density part
you specify the parameter density_density as
density_density = True
Note
It can not be used in conjunction to the Hubbard-I solver or the double-counting correction.
local potential¶
An arbitrary local potential can be implemented using parameters local_potential_*.
The format looks like
[model]
local_potential_matrix = {0: 'pot0.txt', 1: 'pot1.txt'}
local_potential_factor = 0.01
Here, local_potential_matrix describes, in the python dictionary format, a set of the inequivalent shell index ish and the filename which defines the local potential matrix.
The parameter local_potential_factor defines a prefactor to the potential matrix.
For example, the Zeeman term along z-axis for S=1/2 is represented by
$ cat pot0.txt
# spin orb1 orb2 Re Im
0 0 0 0.5 0.
1 0 0 -0.5 0.
and the magnetic field is specified by local_potential_factor.
[system] block¶
This block includes thermodynamic parameters and some technical parameters such as the number of Matsubara frequencies.
Name |
Type |
Default |
Description |
|---|---|---|---|
beta |
Float |
1.0 |
Inverse temperature. This parameter is overridden, if T is given. |
T |
Float |
-1.0 |
Temperature. If this parameter is given, beta is overridden by 1/T. |
n_iw |
Integer |
2048 |
Number of Matsubara frequencies |
fix_mu |
Bool |
False |
Whether or not to fix chemical potential to a given value. |
mu |
Float |
0.0 |
Initial chemical potential. |
prec_mu |
Float |
0.0001 |
Threshold for calculating chemical potential with the bisection method. |
with_dc |
Bool |
False |
Whether or not use double-counting correction (See below) |
dc_type |
String |
HF_DFT |
Chosen from ‘HF_DFT’ (default), ‘HF_imp’, ‘FLL’ |
If the parameter with_dc is specified to True,
the following part of the self-energy is subtracted to avoid the double-counting error of
the self-energy.
where \(\langle \cdots \rangle_0\) indicates the expectation value at the initial (Kohn-Sham) state.
[impurity_solver] block¶
This block specifies an impurity solver to be used and necessary parameters for running the solver program.
Name |
Type |
Default |
Description |
|---|---|---|---|
name |
String |
null |
Name of impurity solver. Available options are null, TRIQS/cthyb, TRIQS/hubbard-I, ALPS/cthyb, ALPS/cthyb-seg, pomerol. |
basis_rotation |
String |
None |
You can specify either ‘Hloc’, ‘None’, or the location of a file.. |
Additionally, we have to specify solver-dependent parameters in the way like n_cycles{int} = 500000.
For details, see the reference manual for each solver.
[control] block¶
This block includes parameters that control the self-consistency loop of DMFT.
Name |
Type |
Default |
Description |
|---|---|---|---|
max_step |
Integer |
100 |
Maximum steps of DMFT loops |
sigma_mix |
Float |
0.5 |
Mixing parameter for self-energy |
restart |
Bool |
False |
Whether or not restart from a previous calculation stored in a HDF file. |
initial_static_self_energy |
String |
None |
dict of {ish: ‘filename’} to specify initial value of the self-energy of ish-th shell. The file format is the same as local_potential_matrix. |
initial_self_energy |
String |
None |
Filename containing initial self-energy in the same format as sigma.dat generated by dcore_check. |
time_reversal |
Bool |
False |
If true, an average over spin components are taken. |
symmetry_generators |
String |
None |
Generators for symmetrization of self-energy. |
n_converge |
Integer |
1 |
The DMFT loop is terminated if the convergence criterion defined with converge_tol is satisfied n_converge times consecutively. |
converge_tol |
Float |
0.0 |
Tolerance in the convergence check. The chemical potential and the renormalization factor are examined. |
[tool] block¶
This block includes parameters that are solely used by dcore_post.
Name |
Type |
Default |
Description |
|---|---|---|---|
nnode |
Integer |
0 |
[NOT USED] Number of node for the k path |
nk_line |
Integer |
8 |
Number of k along each line |
knode |
String |
[(G,0.0,0.0,0.0),(X,1.0,0.0,0.0)] |
The name and the fractional coordinate of each k-node. |
omega_min |
Float |
-1.0 |
Minimum value of real frequency |
omega_max |
Float |
1.0 |
Max value of real frequency |
Nomega |
Integer |
100 |
Number of real frequencies |
broadening |
Float |
0.0 |
An additional Lorentzian broadening |
eta |
Float |
0.0 |
Imaginary frequency shift for the Pade approximation |
omega_pade |
Float |
1e+20 |
Cutoff frequency for the Pade approximation. Data in [-i omega_pade, i omega_pade] is used. |
n_pade_min |
Integer |
20 |
Minimum number of Matsubara frequencies used for Pade approximation. |
n_pade_max |
Integer |
-1 |
Maximum number of Matsubara frequencies used for Pade approximation. If negative, this will be replaced with n_iw in [system] block. |
omega_check |
Float |
0.0 |
Maximum frequency for dcore_check. If not specified, a fixed number of Matsubara points are taken. |
nk_mesh |
Integer |
0 |
Number of k points along each axis for computation of A(k,omega) on a 3D mesh |
nk0_mesh |
Integer |
0 |
Number of k points along b_0 for computation of A(k,omega) on a 3D mesh |
nk1_mesh |
Integer |
0 |
Number of k points along b_1 for computation of A(k,omega) on a 3D mesh |
nk2_mesh |
Integer |
0 |
Number of k points along b_2 for computation of A(k,omega) on a 3D mesh |
[mpi] block¶
This block includes parameters which are read by dcore and dcore_post.
Name |
Type |
Default |
Description |
|---|---|---|---|
command |
String |
mpirun -np # |
Command for executing a MPI job. # will be relaced by the number of processes. |
When an option -DMPIEXEC=<MPIRUN> is passed to the cmake command,
The default value of command will be replaced with <MPIRUN>.