4.1. 多元系イオン結晶 MgAl2O4 の, Mg, Al 原子の反転度計算¶
このチュートリアルの入力ファイルは examples/standard/spinel にあります。
4.1.1. QuantumESPRESSO を用いた例¶
abICS の入力ファイルの準備¶
abICS の入力ファイルは input_qe.toml です。
[replica]
nreplicas = 2
nprocs_per_replica = 1
kTstart = 1000.0
kTend = 1200.0
nsteps = 5 # Number of steps for sampling
RXtrial_frequency = 1
sample_frequency = 1
print_frequency = 1
交換モンテカルロ法に関する情報です。 この例では簡単のため、レプリカ数を2つに設定し、全ステップ数は 5 にしてあります。
[solver]
type = 'qe'
path= './pw.x'
base_input_dir = './baseinput'
perturb = 0.0
run_scheme = 'mpi_spawn'
第一原理エネルギー計算を行うソルバーの情報です。
この例では QuantumESPRESSO (QE) を用いるために type を 'qe' にしています。
QE のエネルギー計算ソルバー pw.x が、実行箇所にあることを path で指定しています(シンボリックリンクでも構いません)。
ソルバーごとに固有の入力パラメータファイルが収められたディレクトリとして、 base_input_dir を用いて ./baseinput を指定しています。
perturb は構造最適化のために原子位置を乱数でずらすパラメータですが、今回は構造最適化を行わないために 0 にしてあります。
abICS は pw.x を MPI_Comm_spawn で起動するため、 run_scheme として mpi_spawn を与えます。
[config]
unitcell = [[8.1135997772, 0.0000000000, 0.0000000000],
[0.0000000000, 8.1135997772, 0.0000000000],
[0.0000000000, 0.0000000000, 8.1135997772]]
supercell = [1,1,1]
[config] は原子位置を指定するセクションです。
unitcell および supercell でシミュレーションする空間を(オングストローム単位で)指定します。
[[config.base_structure]]
type = "O"
coords = [
[0.237399980, 0.237399980, 0.237399980],
[0.762599945, 0.762599945, 0.762599945],
[0.512599945, 0.012600004, 0.737399936],
[0.487399966, 0.987399936, 0.262599975],
[0.012600004, 0.737399936, 0.512599945],
[0.987399936, 0.262599975, 0.487399966],
[0.737399936, 0.512599945, 0.012600004],
[0.262599975, 0.487399966, 0.987399936],
[0.987399936, 0.487399966, 0.262599975],
[0.012600004, 0.512599945, 0.737399936],
[0.487399966, 0.262599975, 0.987399936],
[0.512599945, 0.737399936, 0.012600004],
[0.262599975, 0.987399936, 0.487399966],
[0.737399936, 0.012600004, 0.512599945],
[0.237399980, 0.737399936, 0.737399936],
[0.762599945, 0.262599975, 0.262599975],
[0.512599945, 0.512599945, 0.237399980],
[0.487399966, 0.487399966, 0.762599945],
[0.012600004, 0.237399980, 0.012600004],
[0.987399936, 0.762599945, 0.987399936],
[0.987399936, 0.987399936, 0.762599945],
[0.012600004, 0.012600004, 0.237399980],
[0.487399966, 0.762599945, 0.487399966],
[0.512599945, 0.237399980, 0.512599945],
[0.737399936, 0.237399980, 0.737399936],
[0.262599975, 0.762599945, 0.262599975],
[0.237399980, 0.512599945, 0.512599945],
[0.762599945, 0.487399966, 0.487399966],
[0.762599945, 0.987399936, 0.987399936],
[0.237399980, 0.012600004, 0.012600004],
[0.737399936, 0.737399936, 0.237399980],
[0.262599975, 0.262599975, 0.762599945],
]
[[config.base_structure]] は、モンテカルロ計算で動かさない原子を指定するセクションです。
type で原子名を指定し、 coords でその原子が位置する分率座標を指定します。
coords ではリストのリストだけではなく、空白区切り3列で、原子数だけ行があるような数値行列を表す文字列を与えることもできます。
[[config.defect_structure]]
coords = [
[0.000000000, 0.000000000, 0.000000000],
[0.749999940, 0.249999985, 0.499999970],
[0.249999985, 0.749999940, 0.499999970],
[0.249999985, 0.499999970, 0.749999940],
[0.749999940, 0.499999970, 0.249999985],
[0.499999970, 0.749999940, 0.249999985],
[0.499999970, 0.249999985, 0.749999940],
[0.000000000, 0.499999970, 0.499999970],
[0.749999940, 0.749999940, 0.000000000],
[0.249999985, 0.249999985, 0.000000000],
[0.249999985, 0.000000000, 0.249999985],
[0.749999940, 0.000000000, 0.749999940],
[0.499999970, 0.000000000, 0.499999970],
[0.000000000, 0.749999940, 0.749999940],
[0.000000000, 0.249999985, 0.249999985],
[0.499999970, 0.499999970, 0.000000000],
[0.374999970, 0.374999970, 0.374999970],
[0.624999940, 0.624999940, 0.624999940],
[0.374999970, 0.874999940, 0.874999940],
[0.624999940, 0.124999993, 0.124999993],
[0.874999940, 0.874999940, 0.374999970],
[0.124999993, 0.124999993, 0.624999940],
[0.874999940, 0.374999970, 0.874999940],
[0.124999993, 0.624999940, 0.124999993],
]
[[config.defect_structure]] はモンテカルロ計算で動かす原子の位置を指定するセクションです。
[[config.defect_structure.groups]]
name = 'Al'
# species = ['Al'] # default
# coords = [[[0,0,0]]] # default
num = 16
[[config.defect_structure.groups]]
name = 'Mg'
# species = ['Mg'] # default
# coords = [[[0,0,0]]] # default
num = 8
[[config.defect_structure.groups]] はモンテカルロ計算で動かす原子の名前と数を指定するセクションです。
Al 原子 16 個と、 Mg 原子 8 個を [[config.defect_structure]] で指定した座標中に動かします。
QE の入力ファイルの準備¶
abICS では、内部で保持した原子座標をもとに QE の入力ファイルを生成しますが、原子座標以外の情報、例えば擬ポテンシャルの情報は、ユーザが与える必要があります。
QE の場合は、この情報は base_input_dir で指定したディレクトリの中に scf.in という名前で、 pw.x で使うファイルをそのまま置きます。
このファイルをもとに、ユニットセルや原子座標などを書き換えた入力ファイルが自動生成されます。
注意点 - 擬ポテンシャルディレクトリ
pseudo_dirは絶対パスで渡す必要があります。~はユーザのホームディレクトリに展開されます。 -disk_ioをnoneにすると計算が進まなくなります。lowにしてください。
実行・解析¶
MPI 実行におけるプロセス数やオプションの設定については「実行方法」を参照してください。
mpiexec -np 2 abics input_qe.toml
abics 計算終了後、反転度(もともと Mg 原子があった位置にAl 原子が占めている割合)を計算します。
そのために examples/standard/spinel ディレクトリにある separateT.py と calc_DOI.py スクリプトを用います。
まず、 separateT.py を使い、レプリカごとに分かれていた構造情報を温度ごとにまとめます:
mpiexec -np 2 python3 ./separateT.py input_qe.toml
結果が Tseparate ディレクトリに格納されます。
このディレクトリが既にあった場合は、 Tseparate.DATE というディレクトリに退避されます(DATE は現在時刻)。
つぎに、 calc_DOI.py スクリプトで、反転度を計算します:
mpiexec -np 2 python3 ./calc_DOI.py input_qe.toml
結果が Tseparate/DOI_T.dat に出力されます。
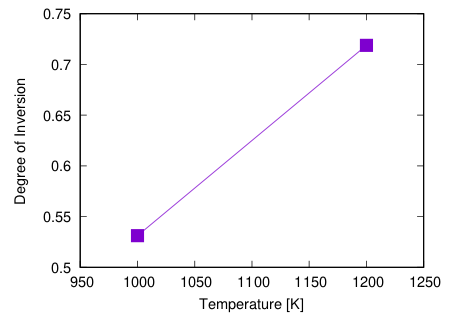
レプリカ数を増やすと様々な温度で計算ができます。
また、ステップ数を増やすことでよりきれいな結果を得られます。
モンテカルロ平均で最初に捨てるサンプル数は calc_DOI.py スクリプトにある throwout の値で指定できます。
例えば、17 レプリカ、20 ステップで計算をすると
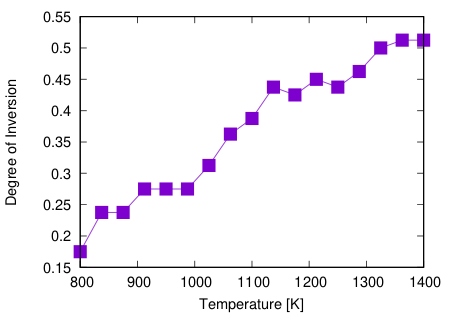
といった結果が得られます。 温度を上げるほど反転度が上がっていくのがわかります。