Calculate diagonal components, i.e. \( H_d |\phi_0> = E_d |\phi_0> \). More...
#include <bitcalc.h>#include "FileIO.h"#include "diagonalcalc.h"#include "mltplySpinCore.h"#include "wrapperMPI.h" Include dependency graph for diagonalcalc.c:
Include dependency graph for diagonalcalc.c:
Go to the source code of this file.
Functions | |
| int | SetDiagonalTETransfer (long unsigned int isite1, double dtmp_V, long unsigned int spin, struct BindStruct *X, double complex *tmp_v0, double complex *tmp_v1) |
| Update the vector by the general one-body diagonal interaction, \( \mu_{i\sigma_1} n_ {i\sigma_1}\). (Using in Time Evolution mode). More... | |
| int | SetDiagonalTEInterAll (long unsigned int isite1, long unsigned int isite2, long unsigned int isigma1, long unsigned int isigma2, double dtmp_V, struct BindStruct *X, double complex *tmp_v0, double complex *tmp_v1) |
| Update the vector by the general two-body diagonal interaction, \( H_{i\sigma_1 j\sigma_2} n_ {i\sigma_1}n_{j\sigma_2}\). (Using in Time Evolution mode). More... | |
| int | SetDiagonalTEChemi (long unsigned int isite1, long unsigned int spin, double dtmp_V, struct BindStruct *X, double complex *tmp_v0, double complex *tmp_v1) |
| Update the vector by the chemical potential \( \mu_{i \sigma_1} n_ {i \sigma_1} \) generated by the commutation relation in terms of the general two-body interaction, \( c_ {i \sigma_1} a_{j\sigma_2}c_ {j \sigma_2}a_ {i \sigma_1} = c_ {i \sigma_1}a_ {i \sigma_1}-c_ {i \sigma_1} a_ {i \sigma_1} c_ {j \sigma_2}a_{j\sigma_2}\) . (Using in Time Evolution mode). More... | |
| int | diagonalcalc (struct BindStruct *X) |
| Calculate diagonal components and obtain the list, list_diagonal. More... | |
| int | diagonalcalcForTE (const int _istep, struct BindStruct *X, double complex *tmp_v0, double complex *tmp_v1) |
| int | SetDiagonalCoulombIntra (long unsigned int isite1, double dtmp_V, struct BindStruct *X) |
| Calculate the components for Coulombintra interaction, \( U_i n_ {i \uparrow}n_{i \downarrow} \). More... | |
| int | SetDiagonalChemi (long unsigned int isite1, double dtmp_V, long unsigned int spin, struct BindStruct *X) |
| Calculate the components for the chemical potential \( \mu_{i \sigma_1} n_ {i \sigma_1} \). More... | |
| int | SetDiagonalCoulombInter (long unsigned int isite1, long unsigned int isite2, double dtmp_V, struct BindStruct *X) |
| Calculate the components for Coulombinter interaction, \( V_{ij} n_ {i}n_{j} \). More... | |
| int | SetDiagonalHund (long unsigned int isite1, long unsigned int isite2, double dtmp_V, struct BindStruct *X) |
| Calculate the components for Hund interaction, \( H_{ij}(n_ {i\uparrow}n_{j\uparrow}+ n_ {i\downarrow}n_{j\downarrow})\). More... | |
| int | SetDiagonalInterAll (long unsigned int isite1, long unsigned int isite2, long unsigned int isigma1, long unsigned int isigma2, double dtmp_V, struct BindStruct *X) |
| Calculate the components for general two-body diagonal interaction, \( H_{i\sigma_1 j\sigma_2} n_ {i\sigma_1}n_{j\sigma_2}\). More... | |
Detailed Description
Calculate diagonal components, i.e. \( H_d |\phi_0> = E_d |\phi_0> \).
- Version
- 2.1
add functions to calculate diagonal components for Time evolution.
- Version
- 0.2
modify functions to calculate diagonal components for general spin.
- Version
- 0.1
Definition in file diagonalcalc.c.
Function Documentation
◆ diagonalcalc()
| int diagonalcalc | ( | struct BindStruct * | X | ) |
Calculate diagonal components and obtain the list, list_diagonal.
- Parameters
-
X [in] Struct to get the information of the diagonal operators.
- Return values
-
-1 fail to calculate diagonal components. 0 succeed to calculate diagonal components.
Definition at line 85 of file diagonalcalc.c.
References cDiagonalCalcFinish, cDiagonalCalcStart, cFileNameCheckChemi, cFileNameCheckCoulombIntra, cFileNameCheckHund, cFileNameCheckInterAll, cFileNameCheckInterU, cFileNameTimeKeep, BindStruct::Check, childfopenMPI(), DefineList::CoulombInter, DefineList::CoulombIntra, cProEndCalcDiag, cProStartCalcDiag, BindStruct::Def, diagonalcalcForTE(), DefineList::EDChemi, DefineList::EDNChemi, DefineList::EDParaChemi, DefineList::EDSpinChemi, DefineList::HundCoupling, CheckList::idim_max, DefineList::InterAll_Diagonal, list_Diagonal, DefineList::NCoulombInter, DefineList::NCoulombIntra, DefineList::NHundCoupling, DefineList::NInterAll_Diagonal, DefineList::ParaCoulombInter, DefineList::ParaCoulombIntra, DefineList::ParaHundCoupling, DefineList::ParaInterAll_Diagonal, SetDiagonalChemi(), SetDiagonalCoulombInter(), SetDiagonalCoulombIntra(), SetDiagonalHund(), SetDiagonalInterAll(), stdoutMPI, and TimeKeeper().
Referenced by CalcSpectrum(), and main().
Here is the call graph for this function: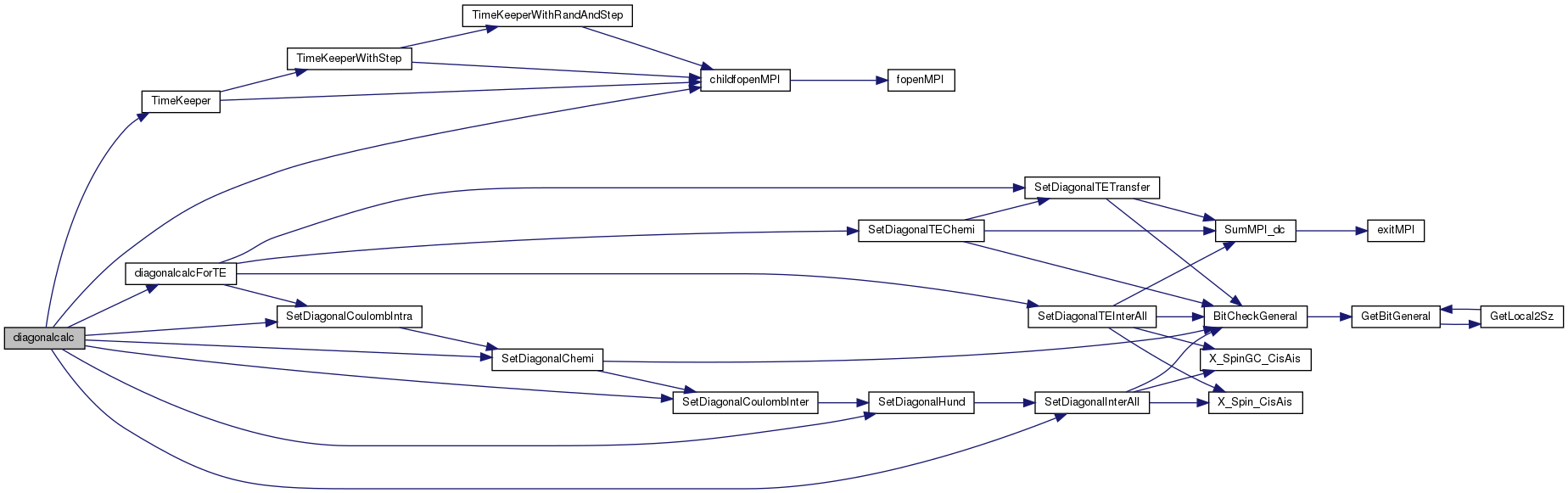 Here is the caller graph for this function:
Here is the caller graph for this function:
◆ diagonalcalcForTE()
| int diagonalcalcForTE | ( | const int | _istep, |
| struct BindStruct * | X, | ||
| double complex * | tmp_v0, | ||
| double complex * | tmp_v1 | ||
| ) |
Definition at line 197 of file diagonalcalc.c.
References BindStruct::Def, DefineList::NTEChemi, DefineList::NTEInterAllDiagonal, DefineList::NTETransferDiagonal, DefineList::ParaTEChemi, DefineList::ParaTEInterAllDiagonal, DefineList::ParaTETransferDiagonal, SetDiagonalCoulombIntra(), SetDiagonalTEChemi(), SetDiagonalTEInterAll(), SetDiagonalTETransfer(), DefineList::SpinTEChemi, DefineList::TEChemi, DefineList::TEInterAllDiagonal, and DefineList::TETransferDiagonal.
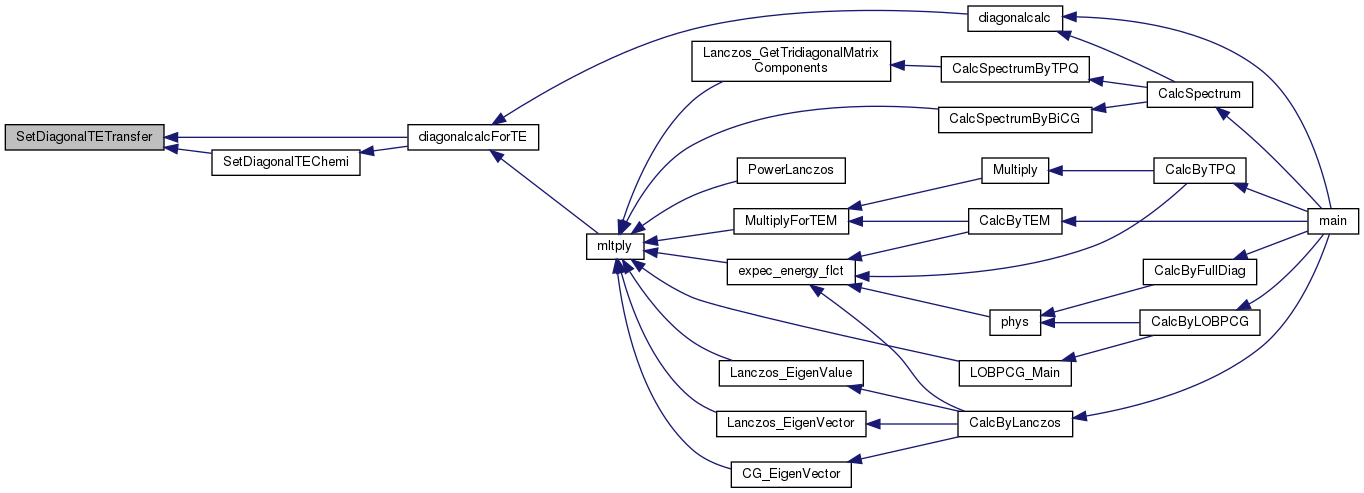
Referenced by diagonalcalc(), and mltply().
Here is the call graph for this function: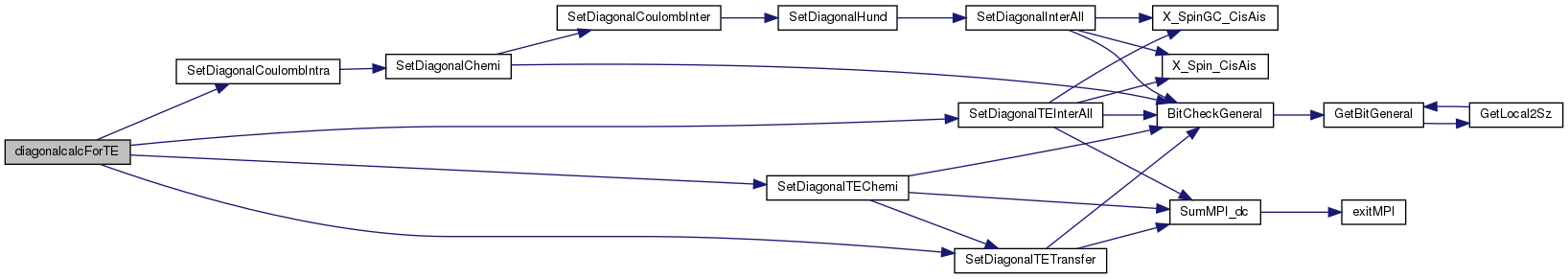 Here is the caller graph for this function:
Here is the caller graph for this function:
◆ SetDiagonalChemi()
| int SetDiagonalChemi | ( | long unsigned int | isite1, |
| double | dtmp_V, | ||
| long unsigned int | spin, | ||
| struct BindStruct * | X | ||
| ) |
Calculate the components for the chemical potential \( \mu_{i \sigma_1} n_ {i \sigma_1} \).
- Parameters
-
isite1 [in] a site number dtmp_V [in] A value of coulombintra interaction \( \mu_{i \sigma_1} \) spin [in] Spin index for the chemical potential X [in] Define list to get dimension number
- Return values
-
-1 fail to calculate the diagonal component. 0 succeed to calculate the diagonal component.
- Version
- 0.1
Definition at line 374 of file diagonalcalc.c.
References BitCheckGeneral(), cErrNoModel, BindStruct::Check, BindStruct::Def, FALSE, DefineList::iCalcModel, CheckList::idim_max, DefineList::iFlgGeneralSpin, list_1, list_Diagonal, myrank, DefineList::Nsite, SetDiagonalCoulombInter(), DefineList::SiteToBit, stdoutMPI, and DefineList::Tpow.
Referenced by diagonalcalc(), and SetDiagonalCoulombIntra().
Here is the call graph for this function: Here is the caller graph for this function:
Here is the caller graph for this function:
◆ SetDiagonalCoulombInter()
| int SetDiagonalCoulombInter | ( | long unsigned int | isite1, |
| long unsigned int | isite2, | ||
| double | dtmp_V, | ||
| struct BindStruct * | X | ||
| ) |
Calculate the components for Coulombinter interaction, \( V_{ij} n_ {i}n_{j} \).
- Parameters
-
isite1 [in] a site number \(i \) isite2 [in] a site number \(j \) dtmp_V [in] A value of coulombinter interaction \( V_{ij} \) X [in] Define list to get the operator information.
- Return values
-
-1 fail to calculate the diagonal component. 0 succeed to calculate the diagonal component.
- Version
- 0.1
Definition at line 544 of file diagonalcalc.c.
References cErrNoModel, BindStruct::Check, BindStruct::Def, DefineList::iCalcModel, CheckList::idim_max, list_1, list_Diagonal, myrank, DefineList::Nsite, SetDiagonalHund(), stdoutMPI, and DefineList::Tpow.
Referenced by diagonalcalc(), and SetDiagonalChemi().
Here is the call graph for this function: Here is the caller graph for this function:
Here is the caller graph for this function:
◆ SetDiagonalCoulombIntra()
| int SetDiagonalCoulombIntra | ( | long unsigned int | isite1, |
| double | dtmp_V, | ||
| struct BindStruct * | X | ||
| ) |
Calculate the components for Coulombintra interaction, \( U_i n_ {i \uparrow}n_{i \downarrow} \).
- Parameters
-
isite1 [in] a site number dtmp_V [in] A value of coulombintra interaction \( U_i \) X [in] Define list to get dimension number
- Return values
-
-1 fail to calculate the diagonal component. 0 succeed to calculate the diagonal component.
- Version
- 0.1
Definition at line 261 of file diagonalcalc.c.
References cErrNoModel, BindStruct::Check, BindStruct::Def, DefineList::iCalcModel, CheckList::idim_max, list_1, list_Diagonal, myrank, DefineList::Nsite, SetDiagonalChemi(), stdoutMPI, and DefineList::Tpow.
Referenced by diagonalcalc(), and diagonalcalcForTE().
Here is the call graph for this function: Here is the caller graph for this function:
Here is the caller graph for this function:
◆ SetDiagonalHund()
| int SetDiagonalHund | ( | long unsigned int | isite1, |
| long unsigned int | isite2, | ||
| double | dtmp_V, | ||
| struct BindStruct * | X | ||
| ) |
Calculate the components for Hund interaction, \( H_{ij}(n_ {i\uparrow}n_{j\uparrow}+ n_ {i\downarrow}n_{j\downarrow})\).
- Parameters
-
isite1 [in] a site number \(i \) isite2 [in] a site number \(j \) dtmp_V [in] A value of Hund interaction \( H_{ij} \) X [in] Define list to get the operator information.
- Return values
-
-1 fail to calculate the diagonal component. 0 succeed to calculate the diagonal component.
- Version
- 0.1
Definition at line 785 of file diagonalcalc.c.
References cErrNoModel, BindStruct::Check, BindStruct::Def, DefineList::iCalcModel, CheckList::idim_max, list_1, list_Diagonal, myrank, DefineList::Nsite, SetDiagonalInterAll(), stdoutMPI, and DefineList::Tpow.
Referenced by diagonalcalc(), and SetDiagonalCoulombInter().
Here is the call graph for this function: Here is the caller graph for this function:
Here is the caller graph for this function:
◆ SetDiagonalInterAll()
| int SetDiagonalInterAll | ( | long unsigned int | isite1, |
| long unsigned int | isite2, | ||
| long unsigned int | isigma1, | ||
| long unsigned int | isigma2, | ||
| double | dtmp_V, | ||
| struct BindStruct * | X | ||
| ) |
Calculate the components for general two-body diagonal interaction, \( H_{i\sigma_1 j\sigma_2} n_ {i\sigma_1}n_{j\sigma_2}\).
- Parameters
-
isite1 [in] a site number \(i \) isite2 [in] a site number \(j \) isigma1 [in] a spin index at \(i \) site. isigma2 [in] a spin index at \(j \) site. dtmp_V [in] A value of general two-body diagonal interaction \( H_{i\sigma_1 j\sigma_2} \) X [in] Define list to get the operator information.
- Return values
-
-1 fail to calculate the diagonal component. 0 succeed to calculate the diagonal component.
- Version
- 0.1
Definition at line 1110 of file diagonalcalc.c.
References BitCheckGeneral(), cErrNoModel, BindStruct::Check, BindStruct::Def, FALSE, DefineList::iCalcModel, CheckList::idim_max, DefineList::iFlgGeneralSpin, list_1, list_Diagonal, myrank, DefineList::Nsite, DefineList::SiteToBit, stdoutMPI, DefineList::Tpow, X_Spin_CisAis(), and X_SpinGC_CisAis().
Referenced by diagonalcalc(), and SetDiagonalHund().
Here is the call graph for this function: Here is the caller graph for this function:
Here is the caller graph for this function:
◆ SetDiagonalTEChemi()
| int SetDiagonalTEChemi | ( | long unsigned int | isite1, |
| long unsigned int | spin, | ||
| double | dtmp_V, | ||
| struct BindStruct * | X, | ||
| double complex * | tmp_v0, | ||
| double complex * | tmp_v1 | ||
| ) |
Update the vector by the chemical potential \( \mu_{i \sigma_1} n_ {i \sigma_1} \)
generated by the commutation relation in terms of the general two-body interaction,
\( c_ {i \sigma_1} a_{j\sigma_2}c_ {j \sigma_2}a_ {i \sigma_1} = c_ {i \sigma_1}a_ {i \sigma_1}-c_ {i \sigma_1} a_ {i \sigma_1} c_ {j \sigma_2}a_{j\sigma_2}\) . (Using in Time Evolution mode).
- Parameters
-
isite1 [in] a site number spin [in] a spin number dtmp_V [in] A value of coulombintra interaction \( \mu_{i \sigma_1} \) X [in] Define list to get dimension number tmp_v0 [in,out] Result vector tmp_v1 [in] Input produced vector
- Return values
-
-1 fail to calculate the diagonal component. 0 succeed to calculate the diagonal component.
- Version
- 2.1
Definition at line 1762 of file diagonalcalc.c.
References BitCheckGeneral(), cErrNoModel, BindStruct::Check, BindStruct::Def, FALSE, DefineList::iCalcModel, CheckList::idim_max, DefineList::iFlgGeneralSpin, BindStruct::Large, list_1, myrank, DefineList::Nsite, LargeList::prdct, SetDiagonalTETransfer(), DefineList::SiteToBit, stdoutMPI, SumMPI_dc(), and DefineList::Tpow.
Referenced by diagonalcalcForTE().
Here is the call graph for this function: Here is the caller graph for this function:
Here is the caller graph for this function:
◆ SetDiagonalTEInterAll()
| int SetDiagonalTEInterAll | ( | long unsigned int | isite1, |
| long unsigned int | isite2, | ||
| long unsigned int | isigma1, | ||
| long unsigned int | isigma2, | ||
| double | dtmp_V, | ||
| struct BindStruct * | X, | ||
| double complex * | tmp_v0, | ||
| double complex * | tmp_v1 | ||
| ) |
Update the vector by the general two-body diagonal interaction, \( H_{i\sigma_1 j\sigma_2} n_ {i\sigma_1}n_{j\sigma_2}\).
(Using in Time Evolution mode).
- Parameters
-
isite1 [in] a site number \(i \) isite2 [in] a site number \(j \) isigma1 [in] a spin index at \(i \) site. isigma2 [in] a spin index at \(j \) site. dtmp_V [in] A value of general two-body diagonal interaction \( H_{i\sigma_1 j\sigma_2} \) X [in] Define list to get the operator information. tmp_v0 [in,out] Result vector tmp_v1 [in] Input produced vector
- Return values
-
-1 fail to calculate the diagonal component. 0 succeed to calculate the diagonal component.
- Version
- 2.1
Definition at line 1431 of file diagonalcalc.c.
References BitCheckGeneral(), cErrNoModel, BindStruct::Check, BindStruct::Def, FALSE, DefineList::iCalcModel, CheckList::idim_max, DefineList::iFlgGeneralSpin, BindStruct::Large, list_1, myrank, DefineList::Nsite, LargeList::prdct, DefineList::SiteToBit, stdoutMPI, SumMPI_dc(), DefineList::Tpow, X_Spin_CisAis(), and X_SpinGC_CisAis().
Referenced by diagonalcalcForTE().
Here is the call graph for this function: Here is the caller graph for this function:
Here is the caller graph for this function:
◆ SetDiagonalTETransfer()
| int SetDiagonalTETransfer | ( | long unsigned int | isite1, |
| double | dtmp_V, | ||
| long unsigned int | spin, | ||
| struct BindStruct * | X, | ||
| double complex * | tmp_v0, | ||
| double complex * | tmp_v1 | ||
| ) |
Update the vector by the general one-body diagonal interaction, \( \mu_{i\sigma_1} n_ {i\sigma_1}\).
(Using in Time Evolution mode).
- Parameters
-
isite1 [in] a site number \(i \) dtmp_V [in] A value of general one-body diagonal interaction \( \mu_{i\sigma_1} \) spin [in] a spin index at \(i \) site. X [in] Define list to get the operator information. tmp_v0 [in,out] Result vector tmp_v1 [in] Input produced vector
- Return values
-
-1 fail to calculate the diagonal component. 0 succeed to calculate the diagonal component.
- Version
- 2.1
Definition at line 1940 of file diagonalcalc.c.
References BitCheckGeneral(), cErrNoModel, BindStruct::Check, BindStruct::Def, FALSE, DefineList::iCalcModel, CheckList::idim_max, DefineList::iFlgGeneralSpin, BindStruct::Large, list_1, myrank, DefineList::Nsite, LargeList::prdct, DefineList::SiteToBit, stdoutMPI, SumMPI_dc(), and DefineList::Tpow.
Referenced by diagonalcalcForTE(), and SetDiagonalTEChemi().
Here is the call graph for this function: Here is the caller graph for this function:
Here is the caller graph for this function: