5.4. Input file for tenes¶
File format is TOML format.
The input file has five sections:
parameter,tensor,evolution,observable,correlation.
5.4.1. parameter section¶
Set various parameters that appear in the calculation, such as the number of updates.
This section has five subsections: general, simple_update, full_update,
ctm, random.
parameter.general¶
General parameters for tenes.
Name |
Description |
Type |
Default |
|---|---|---|---|
|
Calculation mode |
String |
|
|
Whether to limit all tensors to real valued ones |
Boolean |
false |
|
Absolute cutoff value for reading operators |
Real |
0.0 |
|
Whether to calculate and save observables |
Boolean |
true |
|
Interval of measurement in finite temperature calculation and time evolution process |
Integer or list of integers |
10 |
|
Directory for saving result such as physical quantities |
String |
"output" |
|
Directory for saving optimized tensors |
String |
"" |
|
Directory for loading initial tensors |
String |
"" |
modeSpecify the calculation mode
"ground state"Search for the ground state of the Hamiltonian
tenes_stdcalculates the imaginary time evolution operator \(U(\tau) = e^{-\tau H}\) from the Hamiltonian \(H\)
"time evolution"Calculate the time evolution of the observables from the initial state
tenes_stdcalculates the time evolution operator \(U(t) = e^{-it H}\) from the Hamiltonian \(H\)
"finite temperature"Calculate the finite temperature expectation values of the observables
tenes_stdcalculates the imaginary time evolution operator \(U(\tau) = e^{-\tau H}\) from the Hamiltonian \(H\)
is_realWhen set to
true, the type of elements of the tensor becomes real.If one complex operator is defined at least, calculation will end in errors before starting.
iszero_tolWhen the absolute value of operator elements loaded is less than
iszero_tol, it is regarded as zero
meaureWhen set to
false, the stages for measuring and saving observables will be skippedElapsed time
time.datis always saved
measure_intervalSpecify the interval of measurement in time evolution process and finite temperature Calculation
Physical quantitites are calculated and saved each after
measure_intervalupdates
outputSave numerical results such as physical quantities to files in this directory
Empty means
"."(current directory)
tensor_saveSave optimized tensors to files in this directory
If empty no tensors will be saved
tensor_loadRead initial tensors from files in this directory
If empty no tensors will be loaded
parameter.simple_update¶
Parameters in the simple update procedure.
Name |
Description |
Type |
Default |
|---|---|---|---|
|
(Imaginary) time step \(\tau\) in (imaginary) time evolution operator |
Real or list of real |
0.01 |
|
Number of simple updates |
Integer or list of integers |
0 |
|
cutoff of the mean field to be considered zero in the simple update |
Real |
1e-12 |
|
Whether the tensor gauge is fixed |
Boolean |
false |
|
Maximum number of iterations for fixing gauge |
Integer |
100 |
|
Convergence criteria of iterations for fixing gauge |
Real |
1e-2 |
tauSpecify the (imaginary) time step \(\tau\) in (imaginary) time evolution operator
tenes_stduses it to calculate the imaginary time evolution operator \(e^{-\tau H}\) from the Hamiltoniantenesuses it to calculate the time of each measurementFor finite temperature calculation, note that the inverse temperature increase \(2\tau\) at a step because \(\rho(\beta + 2\tau) = U(\tau)\rho(\beta)\bar{U}(\tau)\)
When a list is specified, the time step can be changed for each group of time evolution operators
num_stepSpecify the number of simple updates
When a list is specified, the number of simple updates can be changed for each group of time evolution operators
parameter.full_update¶
Parameters in the full update procedure.
Name |
Description |
Type |
Default |
|---|---|---|---|
|
(Imaginary) time step \(\tau\) in (imaginary) time evolution operator |
Real or list of reals |
0.01 |
|
Number of full updates |
Integer or list of integers |
0 |
|
Cutoff of singular values to be considered as zero when computing environment through full updates |
Real |
1e-12 |
|
Cutoff of singular values to be considered as zero when computing the pseudoinverse matrix with full update |
Real |
1e-12 |
|
Convergence criteria for truncation optimization with full update |
Real |
1e-6 |
|
Maximum iteration number for truncation optimization on full updates |
Integer |
100 |
|
Whether the tensor gauge is fixed |
Boolean |
true |
|
Whether the fast full update is adopted |
Boolean |
true |
parameter.ctm¶
Parameters for corner transfer matrices, CTM.
Name |
Description |
Type |
Default |
|---|---|---|---|
|
Bond Dimension of CTM \(\chi\) |
Integer |
4 |
|
Cutoff of singular values to be considered as zero when computing CTM projectors |
Real |
1e-12 |
|
CTM convergence criteria |
Real |
1e-6 |
|
Maximum iteration number of convergence for CTM |
Integer |
100 |
|
Whether to use only the 1/4 corner tensor in the CTM projector calculation |
Boolean |
true |
|
Whether to replace SVD with random SVD |
Boolean |
false |
|
Ratio of the number of the oversampled elements to that of the obtained elements in random SVD method |
Real |
2.0 |
|
Use mean field environment obtained through simple update instead of CTM |
Boolean |
false |
For Tensor renomalization group approach using random SVD, please see the following reference, S. Morita, R. Igarashi, H.-H. Zhao, and N. Kawashima, Phys. Rev. E 97, 033310 (2018) .
parameter.random¶
Parameters for random number generators.
Name |
Description |
Type |
Default |
|---|---|---|---|
|
Seed of the pseudo-random number generator used to initialize the tensor |
Integer |
11 |
Each MPI process has the own seed as seed plus the process ID (MPI rank).
Example¶
[parameter]
[parameter.general]
is_real = true
[parameter.simple_update]
num_step = 100
tau = 0.01
[parameter.full_update]
num_step = 0 # No full update
tau = 0.01
[parameter.ctm]
iteration_max = 10
dimension = 9 # CHI
5.4.2. tensor section¶
Specify the unit cell information (Information of bonds is given in the hamiltonian (tenes_std) and evolution (tenes) sections.).
Unit cell has a shape of a rectangular with the size of Lx times Ly.
lattice section has an array of subsections unitcell .
Name |
Description |
Type |
Default |
|---|---|---|---|
|
Unit cell size |
Integer or a list of integer |
– |
|
Shift value in skew boundary condition |
Integer |
0 |
When a list of two integers is passed as L_sub, the first element gives the value of Lx and the second one does Ly.
A list of three or more elements causes an error.
If L_sub is an integer, both Lx and Ly will have the same value.
Sites in a unit cell are indexed starting from 0. These are arranged in order from the x direction.
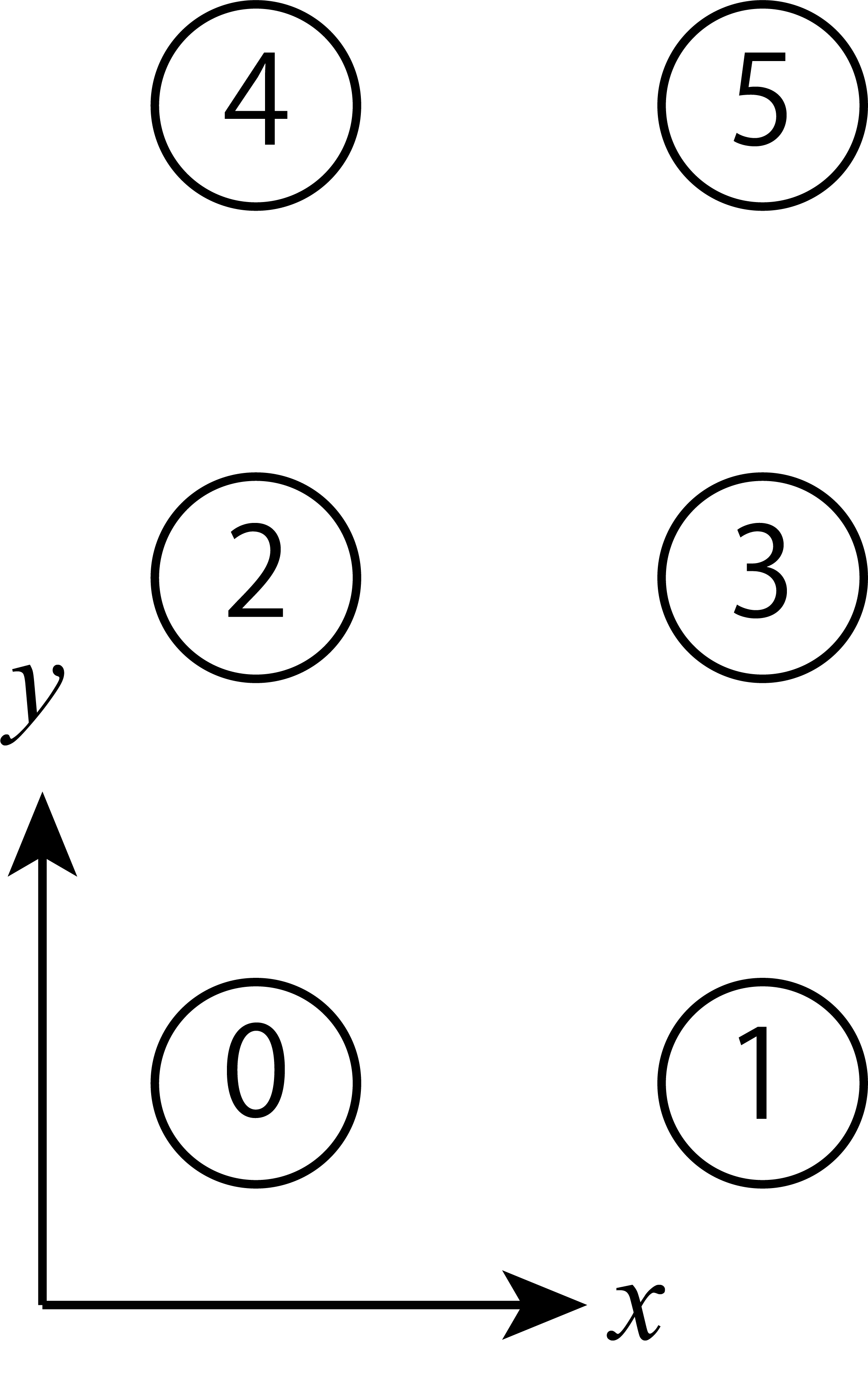
Fig. 5.8 An example for L_sub = [2,3].¶
skew is the shift value in the x direction when moving one unit cell in the y direction.
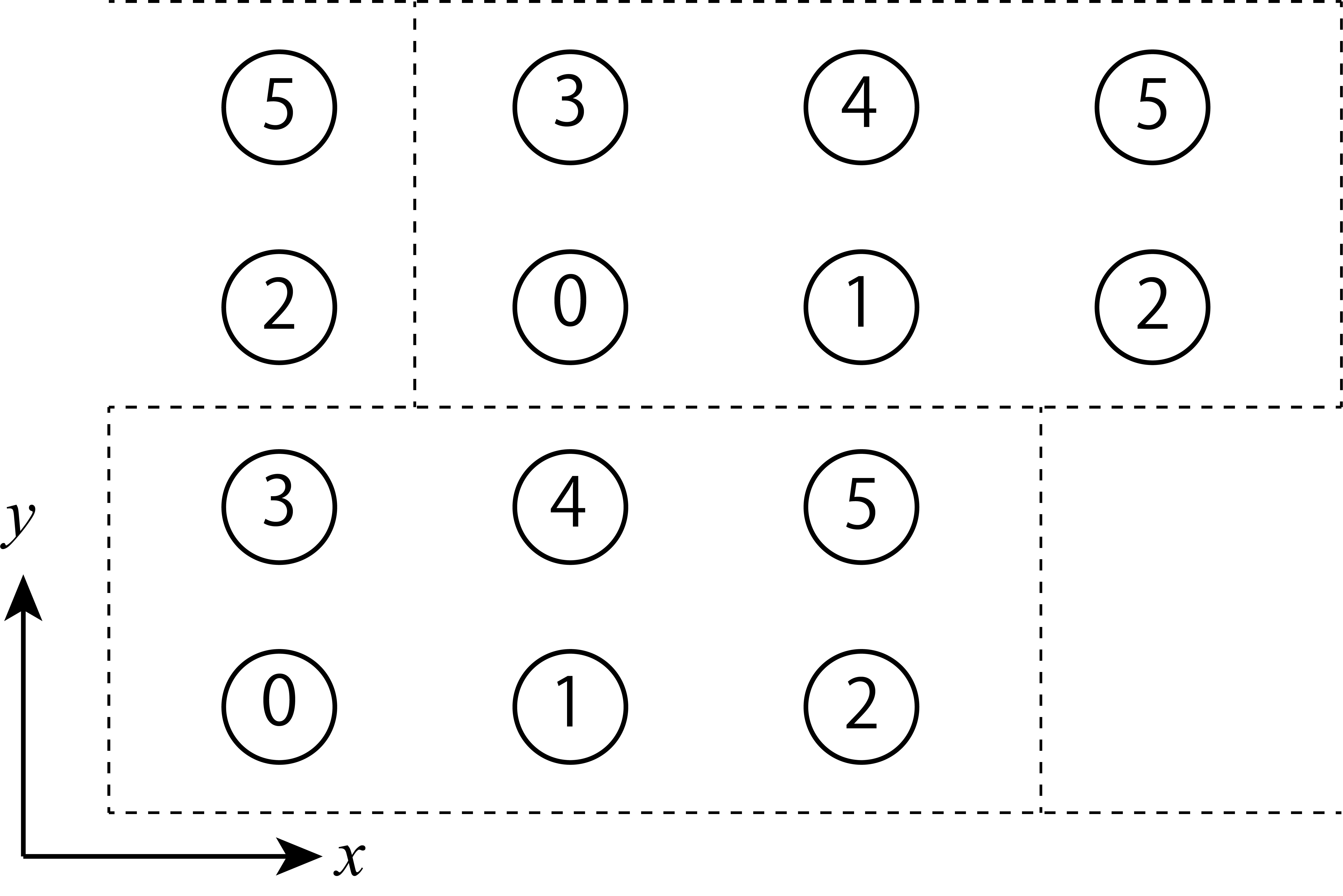
Fig. 5.9 An example for L_sub = [3,2], skew = 1 (ruled line is a separator for unit cell).¶
tensor.unitcell subsection¶
The information of site tensors \(T_{ijkl\alpha}^{(n)}\) is specified. Here, \(i, j, k, l\) indicate the index of the virtual bond, \(\alpha\) indicates the index of the physical bond, and \(n\) indicates the site number.
Name |
Description |
Type |
|---|---|---|
|
Site number |
Integer or a list of integer |
|
Dimension of physical bond for a site tensor |
Integer |
|
Dimension of virtual bonds \(D\) for a site tensor |
Integer or a list of integer |
|
Initial tensor |
a list of real |
|
Noise for initial tensor |
Real |
Multiple sites can be specified at once by setting a list to index.
An empty list [] means all sites.
By setting a list to virtual_dim, individual bond dimensions in four directions can be specified.
The order is left (-x), top (+y), right (+x), and bottom (-y).
An initial state of a system \(|\Psi\rangle\) is represented as the direct product state of the initial states at each site \(i\), \(|\Psi_i\rangle\):
where \(|\Psi_i\rangle = \sum_\alpha A_\alpha |\alpha\rangle_i\) is the initial state at \(i\) site.
Site tensors are initialized to realize this product state with some noise.
initial_state specifies (real) values of expansion coefficient \(A_\alpha\), which will be automatically normalized.
The tensor itself is initialized such that all elements with a virtual bond index of 0 are \(T_{0000\alpha} = A_\alpha\).
The other elements are independently initialized by a uniform random number of [-noise, noise).
For example, in the case of \(S=1/2\) ,
set initial_state = [1.0, 0.0] when you want to set the initial state as the state \(|\Psi_i\rangle = |\uparrow\rangle = |0\rangle\).
When you want to set the initial state as the state \(|\Psi_i\rangle = \left(|\uparrow\rangle + |\downarrow\rangle\right)/\sqrt{2}\), set initial_state = [1.0, 1.0].
When an array consisting of only zeros is passed as initil_state, all the elements of the initial tensor will be initialized independently by uniform random value [-noise, noise) .
5.4.3. observable section¶
Define various settings related to physical quantity measurement.
This section has two types of subsections, onesite and twosite.
observable.onesite¶
Define one-body operators that indicate physical quantities defined at each site \(i\).
Name |
Description |
type |
|---|---|---|
|
Operator name |
String |
|
Identification number of operators |
Integer |
|
Site number |
Integer or a list of integer |
|
Dimension of an operator |
Integer |
|
Non-zero elements of an operator |
String |
|
Coefficient of operator (real part) |
Float |
|
Coefficient of operator (imaginary part) |
Float |
name specifies an operator name.
group specifies an identification number of one-site operators.
sites specifies a site number where an operater acts on.
By using a list, the operators can be defined on the multiple sites at the same time.
An empty list [] means all sites.
dim specifies a dimension of an operator.
elements is a string specifying the non-zero element of an operator.
One element is specified by one line consisting of two integers and two floating-point numbers separated by spaces.
The first two integers are the state numbers before and after the act of the operator, respectively.
The latter two floats indicate the real and imaginary parts of the elements of the operator, respectively.
coeff and coeff_im are real and imaginary parts of the coefficient of the operator, respectively.
If omitted, they are set to 1.0 and 0.0, respectively.
Example¶
As an example, the case of \(S^z\) operator for S=1/2
is explained.
First, set the name to name = "Sz" and the identification number to group = 0.
Next, if the same operator is used at all sites, set sites = [].
Otherwise, for example, if there are sites with different spin length \(S\), specify a specific site number such as sites = [0,1].
The dimension of the operator is dim = 2, because it is the size of the matrix shown above.
Finally, the operator element is defined. When we label two basis on site as \(|\uparrow\rangle = |0\rangle\) and \(|\downarrow\rangle = |1\rangle\), non-zero elements of \(S^z\) are represented as
elements = """
0 0 0.5 0.0
1 1 -0.5 0.0
"""
As a result, \(S^z\) operator for S=1/2 is defined as follows:
[[observable.onesite]]
name = "Sz"
group = 0
sites = []
dim = 2
elements = """
0 0 0.5 0.0
1 1 -0.5 0.0
"""
observable.twosite¶
Define two-body operators that indicate physical quantities defined on two sites.
Name |
Description |
Type |
|---|---|---|
|
Operator name |
String |
|
Identification number of operators |
Integer |
|
Bond |
String |
|
Dimension of an operator |
Integer |
|
Non-zero elements of an operator |
String |
|
Index of onesite operators |
A list of integer |
|
Coefficient of operator (real part) |
Float |
|
Coefficient of operator (imaginary part) |
Float |
name specifies an operator name.
group specifies an identification number of two sites operators.
bonds specifies a string representing the set of site pairs on which the operator acts.
One line consisting of three integers means one site pair.
The first integer is the number of the source site.
The last two integers are the coordinates (dx, dy) of the other site (target site) from the source site.
Both dx and dy must be in the range \(-3 \le dx \le 3\).
dim specifies a dimension of an operator.
In other words, the number of possible states of the site where the operator acts on.
In the case of interaction between two \(S=1/2\) spins, for example, dim = [2, 2] .
elements is a string specifying the non-zero element of an operator.
One element consists of one line consisting of four integers and two floating-point numbers separated by spaces.
The first two integers are the status numbers of the source site and target site before the operator acts on.
The next two integers show the status numbers of the source site and target site after the operator acts on.
The last two floats indicate the real and imaginary parts of the elements of the operator.
Using ops, a two-body operator can be defined as a direct product of the one-body operators defined in observable.onesite.
For example, if \(S^z\) is defined as group = 0 in observable.onesite, \(S ^ z_iS ^ z_j\) can be expressed as ops = [0,0].
If both elements and ops are defined, the process will end in error.
coeff and coeff_im are real and imaginary parts of the coefficient of the operator, respectively.
If omitted, they are set to 1.0 and 0.0, respectively.
Example¶
As an example, for the calculation of the energy of the bond Hamiltonian for S=1/2 Heisenberg model on square lattice at Lsub=[2,2] , the way to define two site operators (equal to the Hamiltonian)
is explained below.
First, the name and identification number is set as name = "hamiltonian" and group = 0.
dim = [2,2] because the state of each site is a superposition of the two states
\(|\uparrow\rangle\) and \(|\downarrow\rangle\).
Next, let’s define the bonds. In this case, site indecies are given as shown in bond_22 .
The bond connecting 0 and 1 is represented as 0 1 0 because 1 is located at (1,0) from 0.
Similarly, The bond connecting 1 and 3 is represented as 1 0 1 because 3 is located at (0,1) from 1.
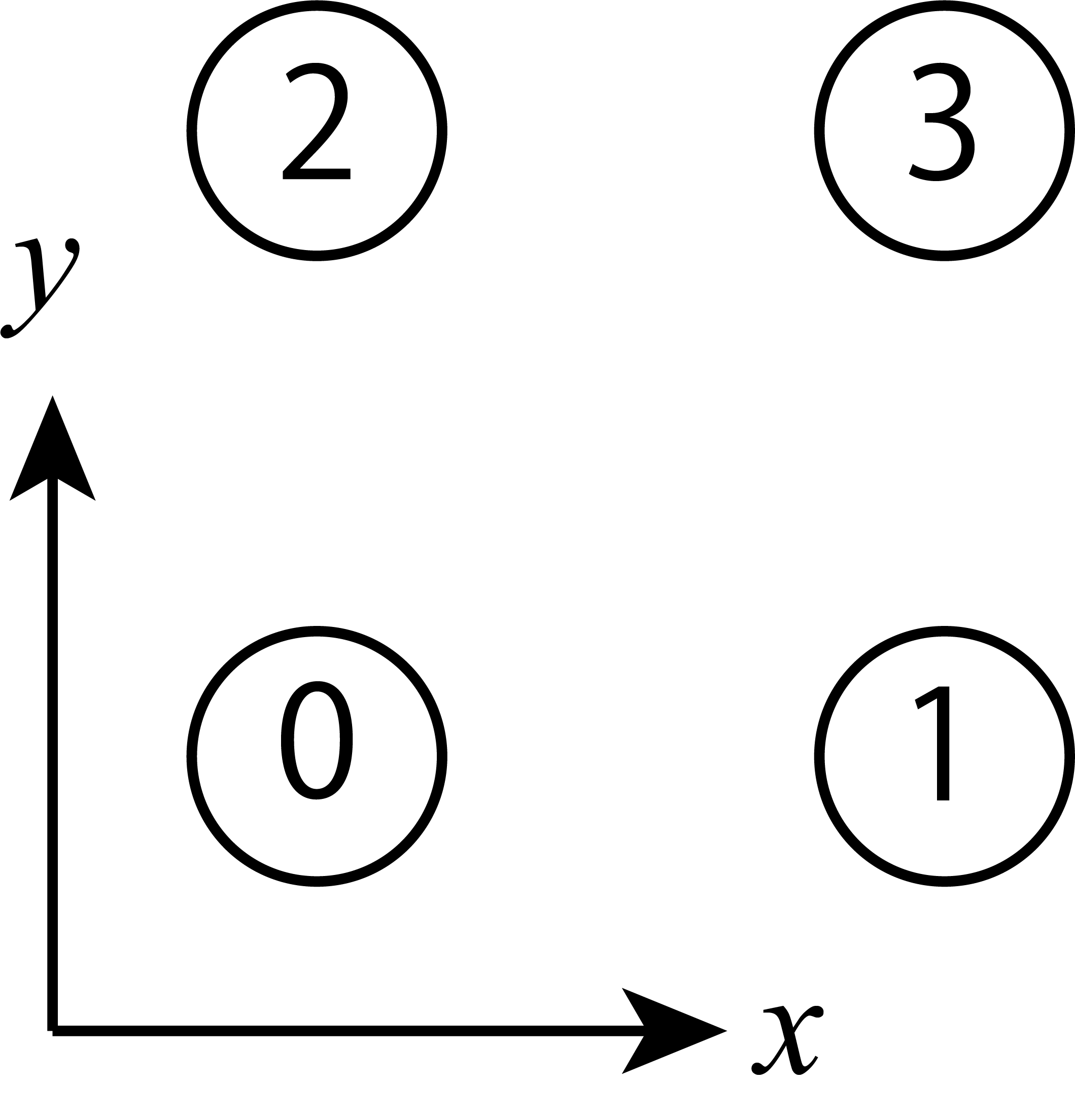
Fig. 5.10 Site indecies of the S=1/2 Heisenberg model on square lattice at Lsub=[2,2] .¶
Finally, how to define the elements of the operator is explained.
First, the basis of the site is needed to be labeled. Here, we label \(|\uparrow\rangle\) as 0 and \(|\downarrow\rangle\) as 1.
Using this basis and label number, for example, one of diagonal elements \(\left\langle \uparrow_i \uparrow_j | \mathcal{H}_{ij} | \uparrow_i \uparrow_j \right\rangle = 1/4\) is specified by 0 0 0 0 0.25 0.0.
Likewise, one of off-diagonal elements \(\left\langle \uparrow_i \downarrow_j | \mathcal{H}_{ij} | \downarrow_i \uparrow_j \right\rangle = 1/2\) is specified by 1 0 0 1 0.5 0.0.
As a result, the Heisenberg Hamiltonian for S=1/2 is defined as follows:
[[observable.twosite]]
name = "hamiltonian"
group = 0
dim = [2, 2]
bonds = """
0 0 1
0 1 0
1 0 1
1 1 0
2 0 1
2 1 0
3 0 1
3 1 0
"""
elements = """
0 0 0 0 0.25 0.0
1 0 1 0 -0.25 0.0
0 1 1 0 0.5 0.0
1 0 0 1 0.5 0.0
0 1 0 1 -0.25 0.0
1 1 1 1 0.25 0.0
"""
observable.multisite¶
Define multi-body operators that indicate physical quantities defined on three or more sites.
It is defined as a direct product of one-body operators defined in observable.onesite.
Name |
Description |
Type |
|---|---|---|
|
Operator name |
String |
|
Identification number of operators |
Integer |
|
Sites |
String |
|
Index of onesite operators |
List of integers |
|
Coefficient of operator (real part) |
Float |
|
Coefficient of operator (imaginary part) |
Float |
name specifies an operator name.
group specifies an identification number of two sites operators.
multisites specifies a string representing the set of sets of sites on which the operator acts.
One line consisting of integers means a set sites.
The first integer is the number of the source site.
The following integers are the coordinates (dx, dy) of the other sites from the source site.
source_site dx2 dy2 dx3 dy3 ... dxN dyNfor N-site operator.All sites must be within a square of size \(4 \times 4\).
Using ops, a multi-body operator can be defined as a direct product of the one-body operators defined in observable.onesite.
For example, if \(S^z\) is defined as group = 0 in observable.onesite, \(S^z_i S^z_j S^z_k\) can be expressed as ops = [0,0,0].
coeff and coeff_im are real and imaginary parts of the coefficient of the operator, respectively.
If omitted, they are set to 1.0 and 0.0, respectively.
5.4.4. evolution section¶
Specify the imaginary time evolution opetrators used in simple and full updates.
One-site and two-sites (nearest neighbor bond) operators can be defined.
This section has two subsections: simple and full.
Name |
Description |
Type |
|---|---|---|
|
Group of the evolution operator |
Integer (0-) |
|
Index of site |
Integer (0-) |
|
Index of source site |
Integer (0-) |
|
Direction from source site to target site |
Integer (0-3) |
|
Dimension of a tensor of imaginary time evolution operator |
A list of integers |
|
Non-zero elements of a tensor of imaginary time evolution operator |
String |
group specifies the group of the evolution operator.
It corresponds to the index of tau and num_steps in parameter.simple_update and parameter.full_update.
site is available for one-site operator, and source_site and source_leg are for two-site operator.
source_leg is specified as an integer from 0 to 3.
Defined as 0: -x, 1: + y, 2: + x, 3: -y in the clockwise order from the -x direction.
dimensions is different from dim in observable section, so you need to specify the dimensions of all legs.
The order of the legs is source_initial, target_initial, source_final, target_final, just like elements.
Example
[evolution]
# One site
[[evolution.simple]]
group = 0
site = 0
dimensions = [2, 2]
elements = """
0 0 1.0012507815756226 0.0
1 1 0.9987507809245809 0.0
"""
# Two site
[[evolution.simple]]
group = 0
source_site = 0
source_leg = 2
dimensions = [2, 2, 2, 2]
elements = """
0 0 0 0 0.9975031223974601 0.0
1 0 1 0 1.0025156589209967 0.0
0 1 1 0 -0.005012536523536871 0.0
1 0 0 1 -0.005012536523536871 0.0
0 1 0 1 1.0025156589209967 0.0
1 1 1 1 0.9975031223974601 0.0
"""
5.4.5. correlation section¶
In this section, the parameters about the site-site correlation function \(C = \left\langle A(\boldsymbol{r}_0)B(\boldsymbol{r}_0 + \boldsymbol{r}) \right\rangle\) is specified. If you omit this section, no correlation functions will be calculated.
Coordinates \(\boldsymbol{r}, \boldsymbol{r}_0\) measured in the system of square lattice TNS. For example, the coordinate of the right neighbor tensor is \(\boldsymbol{r} = (1,0)\) and that of the top neighbor one is \(\boldsymbol{r} = (0,1)\). TeNeS calculates the correlation functions along the positive direction of \(x\) and \(y\) axis, that is,
The coordinate of each site of the unitcell is used as the center coordinate, \(\boldsymbol{r}_0\).
Name |
Description |
Type |
|---|---|---|
|
Maximum distance \(r\) of the correlation function |
Integer |
|
Indices of operators A and B to be measured |
A list of integer |
The operators defined in the observable.onesite section are used.
Example¶
For example, if \(S^z\) is defined as 0th operator and \(S^x\) is defined as 1st one, then \(S^z(0) S^z(r), S^z(0) S^x(r), S^x(0) S^x(r)\) for \(0 \le r \le 5\) are measured by the following definition:
[correlation]
r_max = 5
operators = [[0,0], [0,1], [1,1]]
5.4.6. correlation_length section¶
This section describes how to calculate the correlation length \(\xi\).
Name |
Description |
Type |
Default |
|---|---|---|---|
|
Whether to calculate \(xi\) or not |
Bool |
true |
|
The number of eigenvalues of the transfer matrix to be calculated |
Integer |
4 |
|
Maximum dimension of the transfer matrix where the diagonalization method for dense matrices is used |
Integer |
200 |
|
Dimension of the Hessenberg matrix generated by the Arnoldi method |
Integer |
50 |
|
The number of the initial vectors generated by the restart process of the IRA method |
Integer |
20 |
|
Maximum number of iterations in the IRA method |
Integer |
1 |
|
Relative tolerance used in the Arnoldi method |
Float |
1e-10 |
The correlation length \(\xi\) will be calculated from the dominant eigenvalues of the transfer matrices.
If the dimension of the transfer matrix is less than or equal to maxdim_dense_eigensolver, an eigensolver for dense matrices (LAPACK’s *geev routines) will be used.
If not, an iterative method, the implicit restart Arnoldi method (IRA method), will be used.
In the IRA method, a Hessenberg matrix with the size of arnoldi_maxdim is generated by the Arnoldi process.
Its eigenvalues are approximants of the first arnoldi_maxdim eigenvalues of the original matrix.
If not converged, the IRA method restarts the Arnoldi process with the newly generated arnoldi_restartdim initial vectors.
In the many cases of the transfer matrices, such a process is not necessary (arnoldi_maxiterations = 1).