5.2. Input file for tenes_simple¶
File format is TOML format.
The input file has four sections :
model,parameter,lattice,correlation.The
parametersection is copied to the standard mode input.
5.2.1. model section¶
Specify the model to calculate.
In this version, spin system ("spin") and bosonic system ("boson") are defined.
Name |
Description |
Type |
Default |
|---|---|---|---|
|
Model type (“spin” or “boson”) |
String |
– |
The parameter names such as interactions depend on the model type.
Spin system: "spin"¶
Hamiltonian is described as
The parameters of the one-body terms are defined as follows.
Name |
Description |
Type |
Default |
|---|---|---|---|
|
Magnitude of the local spin |
Real (integer or half integer) |
0.5 |
|
Magnetic field along \(S^x\), \(h^x\) |
Real |
0.0 |
|
Magnetic field along \(S^y\), \(h^y\) |
Real |
0.0 |
|
Magnetic field along \(S^z\), \(h^z\) |
Real |
0.0 |
|
On-site spin anisotropy \(D\) |
Real |
0.0 |
The exchange interaction \(J\) can have a bond dependency.
Name |
Description |
Type |
Default |
|---|---|---|---|
|
Exchange interaction of 0th direction nearest neighbor bond |
Real |
0.0 |
|
Exchange interaction of 1st direction nearest neighbor bond |
Real |
0.0 |
|
Exchange interaction of 2nd direction nearest neighbor bond |
Real |
0.0 |
|
Exchange interaction of 0th direction next nearest neighbor bond |
Real |
0.0 |
|
Exchange interaction of 1st direction next nearest neighbor bond |
Real |
0.0 |
|
Exchange interaction of 2nd direction next nearest neighbor bond |
Real |
0.0 |
|
Exchange interaction of 0th direction third nearest neighbor bond |
Real |
0.0 |
|
Exchange interaction of 1st direction third nearest neighbor bond |
Real |
0.0 |
|
Exchange interaction of 2nd direction third nearest neighbor bond |
Real |
0.0 |
For the next nearest and third nearest neighbor bond, please surround the keyname with the double-quotation marks, ".
The bond direction depends on the lattice defined in the lattice section.
For a square lattice, for example, coupling constants along two bond directions can be defined, x-direction (0) and y-direction (1).
By omitting the direction number, you can specify all directions at once.
You can also specify Ising-like interaction by adding one character of xyz at the end.
If the same bond or component is specified twice or more, an error will occur.
To summarize,
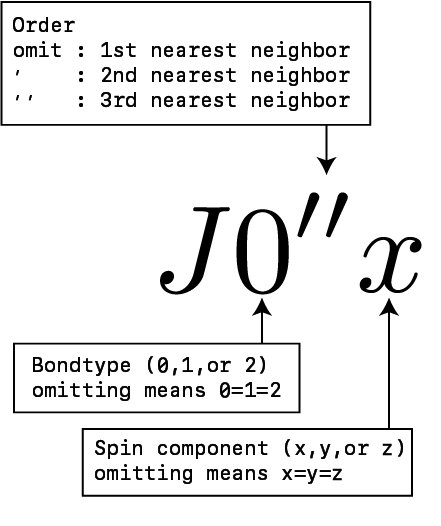
The biquadratic interaction \(B\) can also have a bond dependency like as \(J\).
Name |
Description |
Type |
Default |
|---|---|---|---|
|
Biquadratic interaction of 0th direction nearest neighbor bond |
Real |
0.0 |
|
Biquadratic interaction of 1st direction nearest neighbor bond |
Real |
0.0 |
|
Biquadratic interaction of 2nd direction nearest neighbor bond |
Real |
0.0 |
|
Biquadratic interaction of 0th direction next nearest neighbor bond |
Real |
0.0 |
|
Biquadratic interaction of 1st direction next nearest neighbor bond |
Real |
0.0 |
|
Biquadratic interaction of 2nd direction next nearest neighbor bond |
Real |
0.0 |
|
Biquadratic interaction of 0th direction third nearest neighbor bond |
Real |
0.0 |
|
Biquadratic interaction of 1st direction third nearest neighbor bond |
Real |
0.0 |
|
Biquadratic interaction of 2nd direction third nearest neighbor bond |
Real |
0.0 |
One-site operators \(S ^ z\) and \(S ^ x\) are automatically defined.
If parameter.general.is_real = false, \(S ^ y\) is also defined.
In addition, bond Hamiltonian
and spin correlations on nearest neighbor bonds \(S^\alpha_iS^\alpha_j\) ( \(\alpha=x,y,z\) ) are automatically defined as two-site operators. In the bond Hamiltonian, one body terms (\(h^\alpha\) and \(D\) term) appear only in the nearest neighbor bonds, and \(z\) is the number of the coordinate number.
Bosonic system: "boson"¶
Hamiltonian is described as
where \(b^\dagger\) and \(b\) are the creation and the annihilation operators of a boson, and \(n = b^\dagger b\) is the number operator.
The parameters of the one-body terms are defined as follows.
Name |
Description |
Type |
Default |
|---|---|---|---|
|
Maximum number of particles on a site |
Integer |
1 |
|
Onsite repulsion |
Real |
0.0 |
|
Chemical potential |
Real |
0.0 |
The hopping constant \(t\) and the offsite repulsion \(V\) can have a bond dependency.
Name |
Description |
Type |
Default |
|---|---|---|---|
|
Hopping of 0th direction nearest neighbor bond |
Real |
0.0 |
|
Hopping of 1st direction nearest neighbor bond |
Real |
0.0 |
|
Hopping of 2nd direction nearest neighbor bond |
Real |
0.0 |
|
Hopping of 0th direction next nearest neighbor bond |
Real |
0.0 |
|
Hopping of 1st direction next nearest neighbor bond |
Real |
0.0 |
|
Hopping of 2nd direction next nearest neighbor bond |
Real |
0.0 |
|
Hopping of 0th direction third nearest neighbor bond |
Real |
0.0 |
|
Hopping of 1st direction third nearest neighbor bond |
Real |
0.0 |
|
Hopping of 2nd direction third nearest neighbor bond |
Real |
0.0 |
|
Offsite repulsion of 0th direction nearest neighbor bond |
Real |
0.0 |
|
Offsite repulsion of 1st direction nearest neighbor bond |
Real |
0.0 |
|
Offsite repulsion of 2nd direction nearest neighbor bond |
Real |
0.0 |
|
Offsite repulsion of 0th direction next nearest neighbor bond |
Real |
0.0 |
|
Offsite repulsion of 1st direction next nearest neighbor bond |
Real |
0.0 |
|
Offsite repulsion of 2nd direction next nearest neighbor bond |
Real |
0.0 |
|
Offsite repulsion of 0th direction third nearest neighbor bond |
Real |
0.0 |
|
Offsite repulsion of 1st direction third nearest neighbor bond |
Real |
0.0 |
|
Offsite repulsion of 2nd direction third nearest neighbor bond |
Real |
0.0 |
The bond direction depends on the lattice defined in the lattice section.
For a square lattice, for example, coupling constants along two bond directions can be defined, x-direction (0) and y-direction (1).
By omitting the direction number, you can specify all directions at once.
One-site operators \(n\), \(b\), and \(b^\dagger\) are automatically defined. In addition, bond Hamiltonian
and short range correlations on nearest neighbor bonds \(n_i n_j\), \(b^\dagger_i b_j\), and \(b_i b^\dagger_j\) are automatically defined as two-site operators. In the bond Hamiltonian, one body terms (\(U\) and \(\mu\) term) appear only in the nearest neighbor bonds, and \(z\) is the number of the coordinate number.
5.2.2. lattice section¶
Specify the lattices to calculate. Square, triangular, honeycomb, and Kagome lattices are defined.
Name |
Description |
Type |
Default |
|---|---|---|---|
|
lattice name (square, triangular or honeycomb lattice) |
String |
– |
|
Unit cell size in x direction |
Integer |
– |
|
Unit cell size in y direction |
Integer |
|
|
Bond dimension |
Integer |
– |
|
Inital state |
String |
random |
|
Noise for elements in initial tensor |
Real |
1e-2 |
initial and noise are parameters that determine the initial state of the wave function.
If tensor_load is set in parameter.general, initial is ignored.
initial"ferro": Ferromagnetic stateIn spin system, all sites has \(S^z = S\)
In bosonic system, all sites has \(n = n_{\text{max}}\) particles
"antiferro": Antiferromagnetic stateIn spin system, for square lattice and honeycomb lattice, the Neel order state (\(S^z = S\) for the A sublattice and \(S^z = -S\) for the B sublattice), and for triangular lattice and kagome lattice, the 120 degree order state (spins on sites belonging to the A, B, and C sublattice are pointing to \((\theta, \phi) = (0,0), (2\pi/3, 0)\) and \((2\pi/3, \pi)\) direction, respectively.)
In bosonic system, sites belonging to one sublattice have \(n_\text{max}\) particles and the other sites have no particles.
"random": Random state
noiseThe amount of fluctuation in the elements of the initial tensor
Square lattice¶
A square lattice type = "square lattice" consists of L sites in the \((1,0)\) direction and W sites in the \((0,1)\) direction.
As a concrete example, Fig. 5.1 (a) shows the structure for L=3, W=3.
In addition, the definitions of the first, second and third nearest neighbor bonds are shown in
Fig. 5.1 (b), (c), and (d), respectively.
The blue line represents a bond of bondtype = 0 and the red line represents a bond of bondtype = 1.
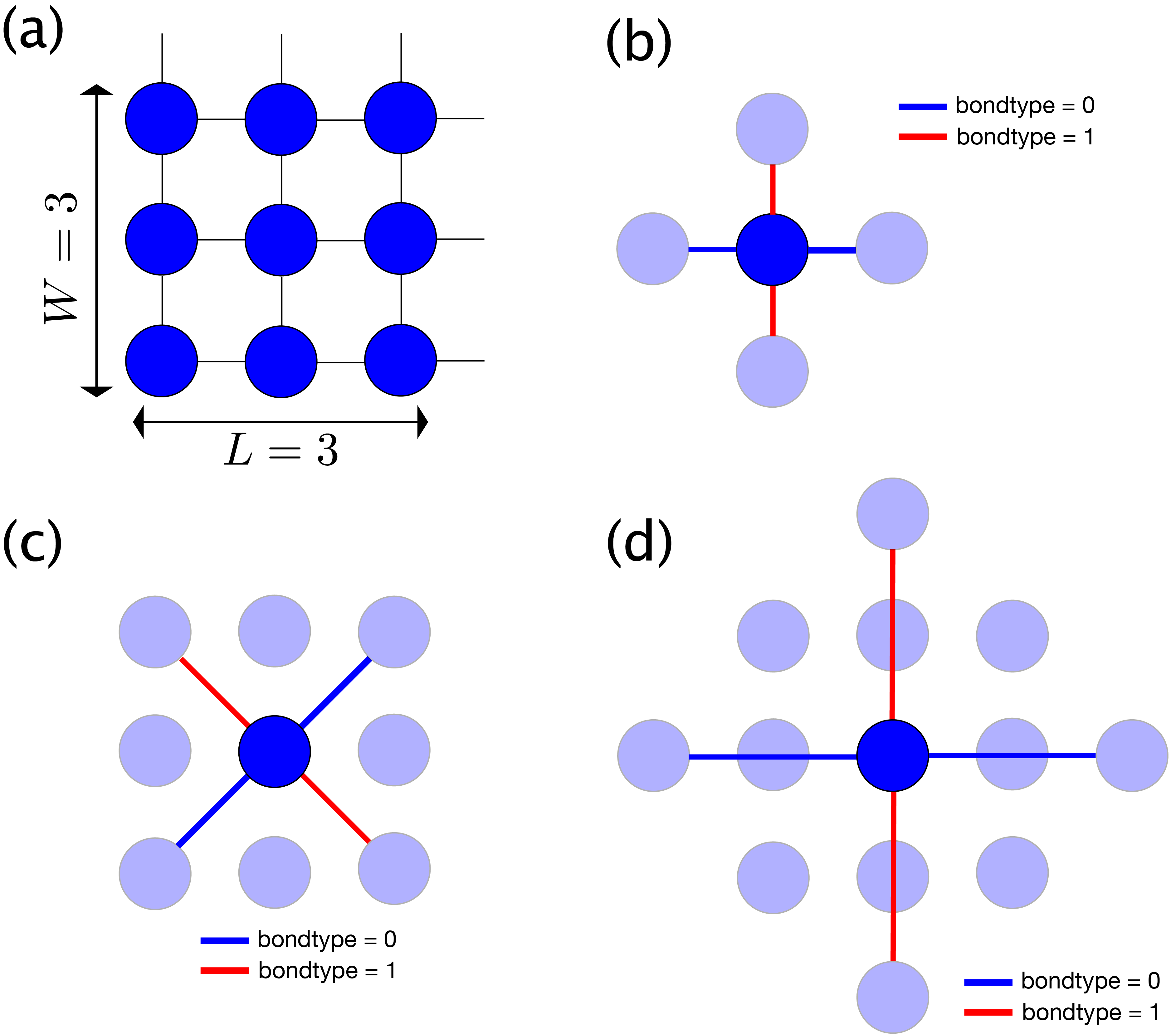
Fig. 5.1 Square lattice.
(a) Site structure with L=3, W=3
(b) Nearest neighbor bonds. bondtype=0 (blue) bond extends in the 0 degree direction and bondtype=1 (red) one in the 90 degree direction.
(c) Second nearest neighbor bonds. bondtype=0 (blue) bond extends in the 45 degree direction and bondtype=1 (red) one in the -45 degree direction.
(d) Third nearest neighbor bonds. bondtype=0 (blue) bond extends in the 0 degree direction and bondtype=1 (red) one in the 90 degree direction.¶
Triangular lattice¶
A triangular lattice type = "triangular lattice" consists of L sites in the \((1,0)\) direction and W sites in the \((1/2, \sqrt{3}/2)\) direction.
As a concrete example, Fig. 5.2 (a) shows the structure for L=3, W=3.
In addition, the definitions of the first, second and third nearest neighbor bonds are shown in
Fig. 5.2 (b), (c), and (d), respectively.
The blue, red, and green lines represent bonds of bondtype = 0, 1, and 2, respectively.
(e) shows the corresponding square TPS with L=3, W=3.
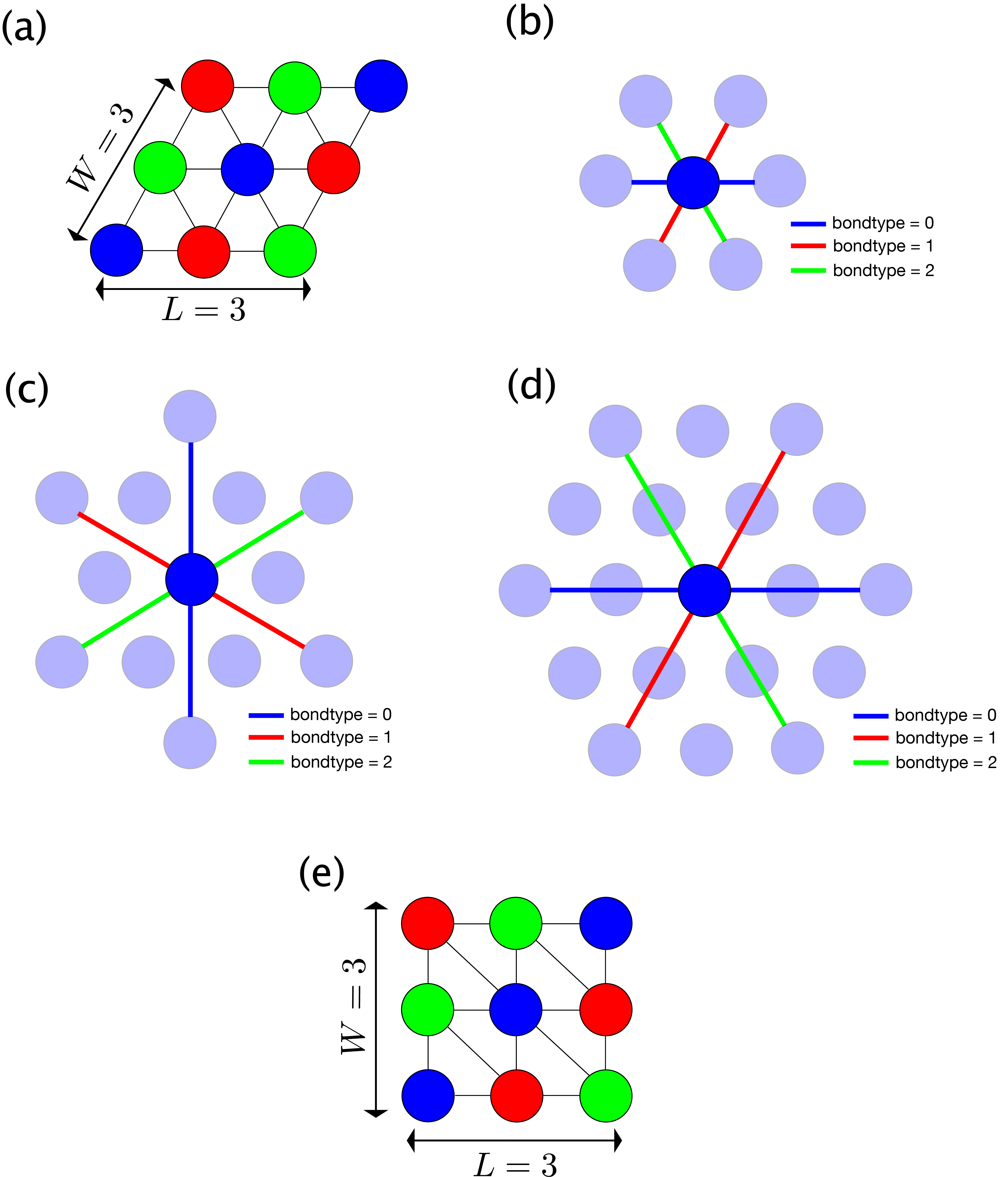
Fig. 5.2 Triangular lattice.
(a) Site structure with L=3, W=3
(b) Nearest neighbor bonds. bondtype=0 (blue) bond extends in the 0 degree direction, bondtype=1 (red) one in the 60 degree direction, and bondtype=2 (green) one in the 120 degree direction.
(c) Second nearest neighbor bonds. bondtype=0 (blue) bond extends in the 90 degree direction, bondtype=1 (red) one in the -30 degree direction, and bondtype=2 (green) one in the 30 degree direction.
(d) Third nearest neighbor bonds. bondtype=0 (blue) bond extends in the 0 degree direction, bondtype=1 (red) one in the 60 degree direction, and bondtype=2 (green) one in the 120 degree direction.
(e) Corrensponding square TPS of the triangular lattice with L=3, W=3.¶
Honeycomb lattice¶
In a honeycomb lattice type = "honeycomb lattice", units consisting of two sites of coordinates \((0, 0)\) and \((\sqrt{3}/2, 1/2)\) are arranged with L units in the \((\sqrt{3},0)\) direction and W units in the \((1/2, 3/2)\) direction.
As a concrete example, Fig. 5.3 (a) shows the structure for L=2, W=2.
In addition, the definitions of the first, second and third nearest neighbor bonds are shown in
Fig. 5.3 (b), (c), and (d), respectively.
The blue, red, and green lines represent bonds of bondtype = 0, 1, and 2, respectively.
(e) shows the corresponding square TPS with L=2, W=2.
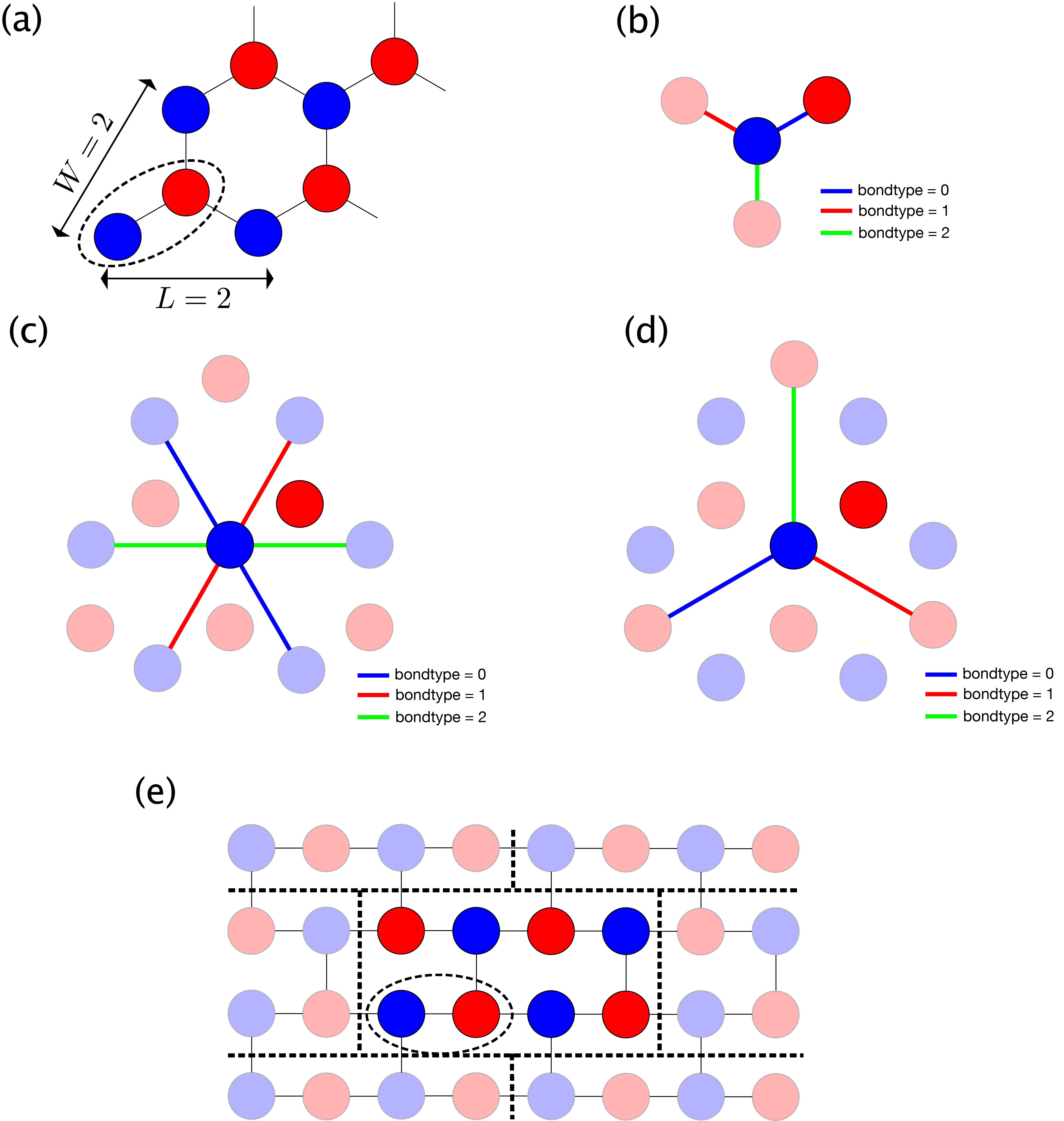
Fig. 5.3 Honeycomb lattice.
(a) Site structure with L=2, W=2. The dashed ellipse denotes one unit.
(b) Nearest neighbor bonds. bondtype=0 (blue) bond extends in the 30 degree direction, bondtype=1 (red) one in the 150 degree direction, and bondtype=2 (green) one in the -90 degree direction.
(c) Second nearest neighbor bonds. bondtype=0 (blue) bond extends in the 120 degree direction, bondtype=1 (red) one in the 60 degree direction, and bondtype=2 (green) one in the 0 degree direction.
(d) Third nearest neighbor bonds. bondtype=0 (blue) bond extends in the -30 degree direction, bondtype=1 (red) one in the -150 degree direction, and bondtype=2 (green) one in the 90 degree direction.
(e) Corresponding square TPS of the honeycomb lattice with L=2, W=2. Note that the most top-right red tensor in the honeycomb lattice moves to the most top-left position, and the boundary condition is skewed.¶
Kagome lattice¶
In a kagome lattice type = "kagome lattice", units consisting of three sites of coordinates \((0, 0)\), \((1, 0)\), and \((1/2, \sqrt{3}/2)\) are arranged with L units in the \((2,0)\) direction and W units in the \((1,\sqrt{3})\) direction.
As a concrete example, Fig. 5.4 (a) shows the structure for L=2, W=2.
In addition, the definitions of the first, second and third nearest neighbor bonds are shown in
Fig. 5.4 (b), (c), and (d), respectively.
The blue and the red lines represent bonds of bondtype = 0, and 1, respectively.
(e) shows the corresponding square TPS with L=2, W=2.
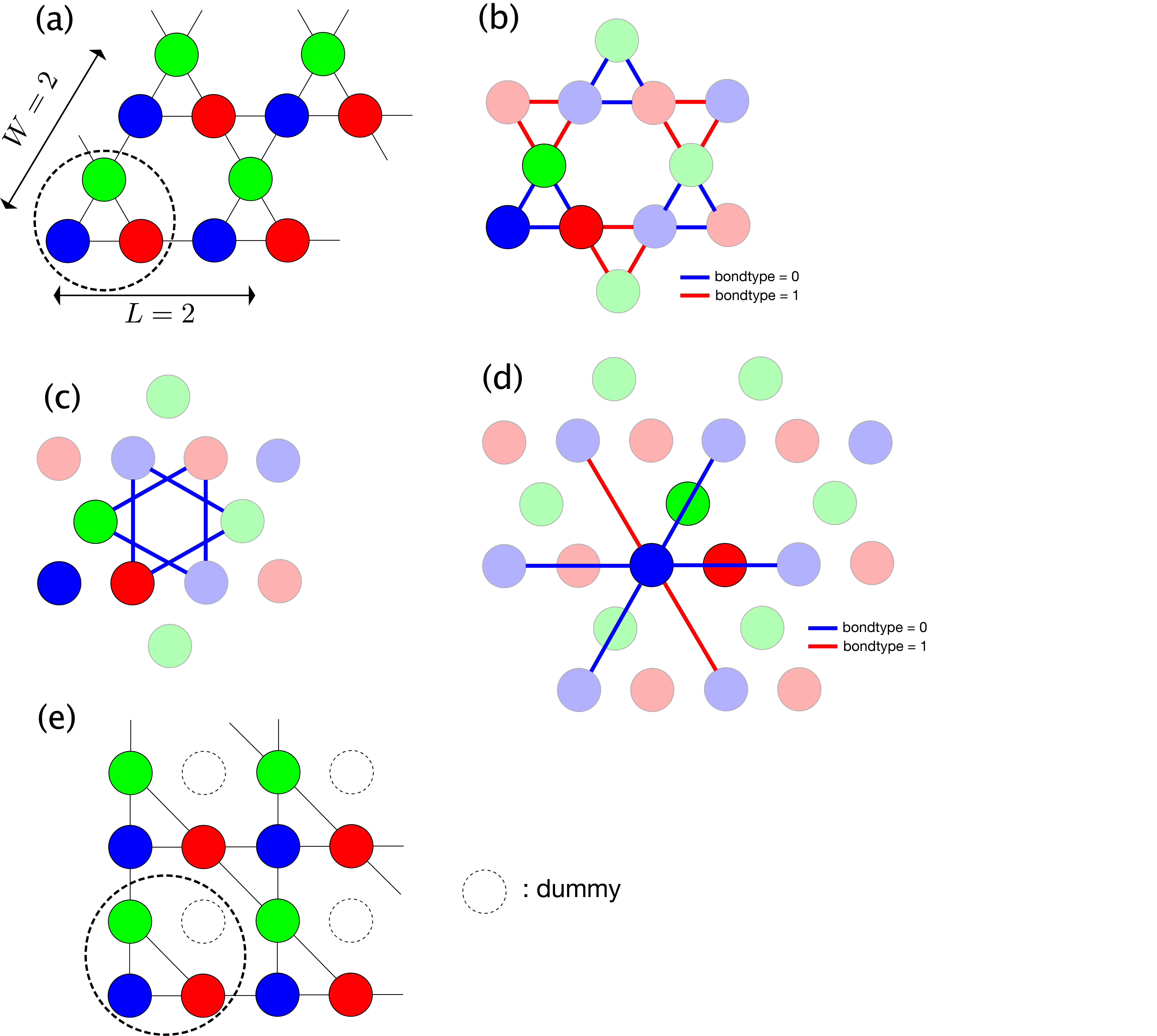
Fig. 5.4 Kagome lattice.
(a) Site structure with L=2, W=2. The dashed circle denotes one unit.
(b) Nearest neighbor bonds. bondtype=0 (blue) bonds form upper triangle and bondtype=1 (red) bonds form lowertriangle.
(c) Second nearest neighbor bonds.
(d) Third nearest neighbor bonds. bondtype=0 (blue) bond passes over a site and bondtype=1 (red) one does not.
(e) Corresponding square TPS of the kagome lattice with L=2, W=2. The white circles are the dummy tensors with bonds of dimension one.¶
5.2.3. parameter section¶
Parameters defined in this section is not used in tenes_simple but they are copied to the input file of tenes_std.
Set various parameters that appear in the calculation, such as the number of updates.
This section has five subsections: general, simple_update, full_update,
ctm, random.
parameter.general¶
General parameters for tenes.
Name |
Description |
Type |
Default |
|---|---|---|---|
|
Calculation mode |
String |
|
|
Whether to limit all tensors to real valued ones |
Boolean |
false |
|
Absolute cutoff value for reading operators |
Real |
0.0 |
|
Whether to calculate and save observables |
Boolean |
true |
|
Interval of measurement in finite temperature calculation and time evolution process |
Integer or list of integers |
10 |
|
Directory for saving result such as physical quantities |
String |
"output" |
|
Directory for saving optimized tensors |
String |
"" |
|
Directory for loading initial tensors |
String |
"" |
modeSpecify the calculation mode
"ground state"Search for the ground state of the Hamiltonian
tenes_stdcalculates the imaginary time evolution operator \(U(\tau) = e^{-\tau H}\) from the Hamiltonian \(H\)
"time evolution"Calculate the time evolution of the observables from the initial state
tenes_stdcalculates the time evolution operator \(U(t) = e^{-it H}\) from the Hamiltonian \(H\)
"finite temperature"Calculate the finite temperature expectation values of the observables
tenes_stdcalculates the imaginary time evolution operator \(U(\tau) = e^{-\tau H}\) from the Hamiltonian \(H\)
is_realWhen set to
true, the type of elements of the tensor becomes real.If one complex operator is defined at least, calculation will end in errors before starting.
iszero_tolWhen the absolute value of operator elements loaded is less than
iszero_tol, it is regarded as zero
meaureWhen set to
false, the stages for measuring and saving observables will be skippedElapsed time
time.datis always saved
measure_intervalSpecify the interval of measurement in time evolution process and finite temperature Calculation
Physical quantitites are calculated and saved each after
measure_intervalupdates
outputSave numerical results such as physical quantities to files in this directory
Empty means
"."(current directory)
tensor_saveSave optimized tensors to files in this directory
If empty no tensors will be saved
tensor_loadRead initial tensors from files in this directory
If empty no tensors will be loaded
parameter.simple_update¶
Parameters in the simple update procedure.
Name |
Description |
Type |
Default |
|---|---|---|---|
|
(Imaginary) time step \(\tau\) in (imaginary) time evolution operator |
Real or list of real |
0.01 |
|
Number of simple updates |
Integer or list of integers |
0 |
|
cutoff of the mean field to be considered zero in the simple update |
Real |
1e-12 |
|
Whether the tensor gauge is fixed |
Boolean |
false |
|
Maximum number of iterations for fixing gauge |
Integer |
100 |
|
Convergence criteria of iterations for fixing gauge |
Real |
1e-2 |
tauSpecify the (imaginary) time step \(\tau\) in (imaginary) time evolution operator
tenes_stduses it to calculate the imaginary time evolution operator \(e^{-\tau H}\) from the Hamiltoniantenesuses it to calculate the time of each measurementFor finite temperature calculation, note that the inverse temperature increase \(2\tau\) at a step because \(\rho(\beta + 2\tau) = U(\tau)\rho(\beta)\bar{U}(\tau)\)
When a list is specified, the time step can be changed for each group of time evolution operators
num_stepSpecify the number of simple updates
When a list is specified, the number of simple updates can be changed for each group of time evolution operators
parameter.full_update¶
Parameters in the full update procedure.
Name |
Description |
Type |
Default |
|---|---|---|---|
|
(Imaginary) time step \(\tau\) in (imaginary) time evolution operator |
Real or list of reals |
0.01 |
|
Number of full updates |
Integer or list of integers |
0 |
|
Cutoff of singular values to be considered as zero when computing environment through full updates |
Real |
1e-12 |
|
Cutoff of singular values to be considered as zero when computing the pseudoinverse matrix with full update |
Real |
1e-12 |
|
Convergence criteria for truncation optimization with full update |
Real |
1e-6 |
|
Maximum iteration number for truncation optimization on full updates |
Integer |
100 |
|
Whether the tensor gauge is fixed |
Boolean |
true |
|
Whether the fast full update is adopted |
Boolean |
true |
parameter.ctm¶
Parameters for corner transfer matrices, CTM.
Name |
Description |
Type |
Default |
|---|---|---|---|
|
Bond Dimension of CTM \(\chi\) |
Integer |
4 |
|
Cutoff of singular values to be considered as zero when computing CTM projectors |
Real |
1e-12 |
|
CTM convergence criteria |
Real |
1e-6 |
|
Maximum iteration number of convergence for CTM |
Integer |
100 |
|
Whether to use only the 1/4 corner tensor in the CTM projector calculation |
Boolean |
true |
|
Whether to replace SVD with random SVD |
Boolean |
false |
|
Ratio of the number of the oversampled elements to that of the obtained elements in random SVD method |
Real |
2.0 |
|
Use mean field environment obtained through simple update instead of CTM |
Boolean |
false |
For Tensor renomalization group approach using random SVD, please see the following reference, S. Morita, R. Igarashi, H.-H. Zhao, and N. Kawashima, Phys. Rev. E 97, 033310 (2018) .
parameter.random¶
Parameters for random number generators.
Name |
Description |
Type |
Default |
|---|---|---|---|
|
Seed of the pseudo-random number generator used to initialize the tensor |
Integer |
11 |
Each MPI process has the own seed as seed plus the process ID (MPI rank).
Example¶
[parameter]
[parameter.general]
is_real = true
[parameter.simple_update]
num_step = 100
tau = 0.01
[parameter.full_update]
num_step = 0 # No full update
tau = 0.01
[parameter.ctm]
iteration_max = 10
dimension = 9 # CHI
5.2.4. correlation section¶
For tenes_simple , correlation functions \(C = \langle A(0)B(r)\rangle\) are not calculated by default.
For calculating correlation functions, they have to be specified in the same file format as the input file of tenes.
For details, See correlation section Input file for tenes.
5.2.5. correlation_length section¶
Parameters defined in this section is not used in tenes_simple but they are copied to the input file of tenes_std.
This section describes how to calculate the correlation length \(\xi\).
Name |
Description |
Type |
Default |
|---|---|---|---|
|
Whether to calculate \(xi\) or not |
Bool |
true |
|
The number of eigenvalues of the transfer matrix to be calculated |
Integer |
4 |
|
Maximum dimension of the transfer matrix where the diagonalization method for dense matrices is used |
Integer |
200 |
|
Dimension of the Hessenberg matrix generated by the Arnoldi method |
Integer |
50 |
|
The number of the initial vectors generated by the restart process of the IRA method |
Integer |
20 |
|
Maximum number of iterations in the IRA method |
Integer |
1 |
|
Relative tolerance used in the Arnoldi method |
Float |
1e-10 |
The correlation length \(\xi\) will be calculated from the dominant eigenvalues of the transfer matrices.
If the dimension of the transfer matrix is less than or equal to maxdim_dense_eigensolver, an eigensolver for dense matrices (LAPACK’s *geev routines) will be used.
If not, an iterative method, the implicit restart Arnoldi method (IRA method), will be used.
In the IRA method, a Hessenberg matrix with the size of arnoldi_maxdim is generated by the Arnoldi process.
Its eigenvalues are approximants of the first arnoldi_maxdim eigenvalues of the original matrix.
If not converged, the IRA method restarts the Arnoldi process with the newly generated arnoldi_restartdim initial vectors.
In the many cases of the transfer matrices, such a process is not necessary (arnoldi_maxiterations = 1).