4.1. ニューラルネットワークの構築¶
ここでは、aenet を用いてニューラルネットワークモデルの構築を行う方法について記載します。
第一原理ソルバーには Quantum ESPRESSO (QE) を使います。
例題としてMgAl2O4スピネルのMg/Al反転度の温度依存性を計算します。
最安定構造では全てのMgが酸素に四面体配位されていて、Alは八面体配位になっています。
このサイト間の反転度の温度依存性をシミュレーションします。
なお、本チュートリアルで使用する入力ファイル一式は examples/active_learning_qe/ にあります。
以下では aenet および GNU parallel のインストールについても簡単に説明していますが、システムに既にインストールされている場合はそちらを使ってください。
なお、計算の実行環境は物性研究所スーパーコンピュータシステムBのohtakaを利用しています。
4.1.1. 事前準備¶
aenetのインストール¶
abICSでは、ニューラルネットワークモデルの構築のためにaenetを利用します。
aenetは http://ann.atomistic.net からダウンロードできます。
Documentation の Installation に従ってインストールしてください。
なお、abICSでは、ニューラルネットワークの学習と評価にaenetの train.x と predict.x を使います。
train.x についてはMPI並列版が利用可能ですが、 predict.x についてはMPIを使用しない実行ファイル(serial)を使用する必要があります。
そのため、makefilesの下にあるserial版もインストールするようにしてください。
GNU parallelのインストール¶
チュートリアルでは、GNU parallelを用いて Quantum Espresso による第一原理計算を並列実行します。 GNU parallelは https://www.gnu.org/software/parallel/ からダウンロードできます (Macの場合はhomebrewにより直接インストールすることも可能です)。 インストールは基本的には、ダウンロードして解凍したディレクトリ に移動した後、
$ ./configure && make && make install
でインストールできます。詳細な設定は公式マニュアルを参考にしてください。
4.1.2. 学習データ生成用入力ファイルの準備¶
ニューラルネットワークの作成にあたり、 原子配置を入力、エネルギーを出力とした学習データを、 第一原理計算により作成する必要があります。 学習データの作成には、
abICS
利用する第一原理ソルバー
に関する入力ファイルを準備する必要があります。
abICS制御ファイル (input.toml)¶
計算対象とする格子構造の定義と、abICSによる能動学習のループ全体の制御、およびレプリカ交換モンテカルロ法に関するパラメータを設定します。
st2abics ツールを使うことで、結晶構造ファイルから input.toml のひな形を自動で生成することができます。
$ cd [example_dir]
$ st2abics st2abics_MgAl2O4.toml MgAl2O4.vasp > input.toml
以下では input.toml の中で第一原理計算によ る学習データに関連するセクションの設定内容をもう少し詳しく解説します。
(i) [mlref] セクション¶
[mlref]
nreplicas = 8
ndata = 5
RXMC計算の結果から、ニューラルネットワークモデルの精度評価と訓練データの拡張のために原子配置を取り出す際のオプションが設定できます。
基本的に、 nreplicas は 後述する [sampling] セクションと同じ値にしてください。
ndata は、RXMC計算で出力される配置の数(後述する [sampling] セクションの nsteps/sample_frequency の値)から、
何個のデータを機械学習用に取り出すかを指定します。従って、RXMC計算で出力される配置の数以下の値に設定してください。
(ii) [mlref.solver] セクション¶
[mlref.solver] # 参照第一原理ソルバーの設定
type = 'qe'
base_input_dir = ['./baseinput_ref', './baseinput_ref', './baseinput_ref'] #, './baseinput_ref']
perturb = 0.05
ignore_species = []
訓練データ(配置エネルギー)の計算に用いるソルバーの設定を行います。この例ではQuantum Espressoで配置エネルギーを求めます。
base_input_dir は自由に設定して構いません。設定したディレクトリの中に、ソルバーの入力ファイルを設置します(後述)。
上記の例のようにリスト形式で複数設定した場合は、各々のディレクトリ内の入力を使った計算が順番に実行されます。 このときに、2番目以降の計算では 前の計算の最終ステップでの構造が初期座標として用いられます。そして、最後の計算のエネルギーが 学習に使われます。例えば、1つ目の入力ファイルでで精度を犠牲にして高速な構造最適化を行い、2番目以降の入力ファイルで 高精度な設定で構造最適化を行うといった ことが可能になります。あるいは、格子ベクトルの緩和を行う場合に、設定した平面波カットオフに基づいて計算メッシュをリセット するために同じ入力の計算を複数回実行するといったことも可能です。
perturb は、ランダムに各原子を変位させることで、対称性を崩した構造から構造最適化を開始するための
設定です。この場合は、構造緩和を行う原子を全て0.05 Å、ランダムな方向に変位させた構造から1番目の計算が開始されます。
ignore_species は、第一原理ソルバーを訓練データ生成に用いる場合は空リストを指定しますが、一部の元素を無視するような
モデルを使って訓練データを生成する場合は、無視する元素を指定します。
(iii) [config] セクション¶
[config] # 以下、結晶格子の情報と、格子上に配置される原子や空孔の情報が続く
unitcell = [[8.1135997772, 0.0000000000, 0.0000000000],
[0.0000000000, 8.1135997772, 0.0000000000],
[0.0000000000, 0.0000000000, 8.1135997772]]
supercell = [1,1,1]
[[config.base_structure]]
type = "O"
coords = [
[0.237399980, 0.237399980, 0.237399980],
[0.762599945, 0.762599945, 0.762599945],
[0.512599945, 0.012600004, 0.737399936],
[0.487399966, 0.987399936, 0.262599975],
...
基本的に st2abics ツールで生成されたものをそのまま利用できます。
モンテカルロサンプリングを行う原子配置の情報が設定されます。
abics_sampling が未実施の場合には、この情報をもとに原子配置がランダムに与えられ、
それらの原子配置を持った第一原理計算用の入力ファイルが生成されます。
abics_sampling が既に実行されている場合には、 config セクションではなく、
モンテカルロサンプリングで与えられた原子配置に対する第一原理計算用の入力ファイルが生成されます。
QE参照ファイルの準備¶
baseinput_ref にQEのscf計算で参照する入力ファイルを配置します。
以下、サンプルディレクトリにある scf.in ファイルを記載します。
&CONTROL
calculation = 'relax'
tstress = .false.
tprnfor = .false.
pseudo_dir = './pseudo'
disk_io = 'low'
wf_collect = .false.
/
&SYSTEM
ecutwfc = 60.0
occupations = "smearing"
smearing = "gauss"
degauss = 0.01
/
&electrons
mixing_beta = 0.7
conv_thr = 1.0d-8
electron_maxstep = 100
/
&ions
/
ATOMIC_SPECIES
Al 26.981 Al.pbe-nl-kjpaw_psl.1.0.0.UPF
Mg 24.305 Mg.pbe-spnl-kjpaw_psl.1.0.0.UPF
O 16.000 O.pbe-n-kjpaw_psl.1.0.0.UPF
ATOMIC_POSITIONS crystal
K_POINTS gamma
なお、擬ポテンシャルを格納したディレクトリ pseudo_dir や
ATOMIC_SPECIES で使用する擬ポテンシャルについて、自分の環境に従い書き換える必要があります。
本サンプルで使用している擬ポテンシャルは以下のリンクからダウンロードできます。
(ダウンロードスクリプト download_pp.sh が用意されています。)
https://pseudopotentials.quantum-espresso.org/upf_files/Al.pbe-nl-kjpaw_psl.1.0.0.UPF
https://pseudopotentials.quantum-espresso.org/upf_files/Mg.pbe-spnl-kjpaw_psl.1.0.0.UPF
https://pseudopotentials.quantum-espresso.org/upf_files/O.pbe-n-kjpaw_psl.1.0.0.UPF
このサンプルでは、QE計算時に構造最適化を行うため calculation = 'relax' を、
計算高速化のため、 K_POINTS は gammma を選択しています。
4.1.3. ニューラルネットワーク生成用入力ファイルの準備¶
本チュートリアルではaenetを用いニューラルネットワークを作成します。 ニューラルネットワークの作成には、
abICS
aenet
に関する入力ファイルを準備・設定する必要があります。
abICS制御ファイル (input.toml)¶
(i) [train] セクション¶
[train] # モデル学習器の設定
type = 'aenet'
base_input_dir = './aenet_train_input'
exe_command = ['generate.x-2.0.4-ifort_serial',
'srun train.x-2.0.4-ifort_intelmpi']
ignore_species = ["O"]
vac_map = []
restart = false
訓練データから配置エネルギー予測モデルを学習する学習器の設定を行います。現在のところ、abICSではaenetのみに
対応しています。 base_input_dir は自由に設定して構いません。設定したディレクトリの中に、学習器の設定ファイルを
設置します(後述)。 exe_command にはaenetの generate.x と train.x へのパスを指定します。
train.x についてはMPI並列版が利用可能で、その場合は、上の例で示すように、MPI実行するためのコマンド
( srun 、 mpirun など)を合わせて設定してください。
ignore_species は、第一原理ソルバーを訓練データ生成に用いる場合は空リストを指定しますが、
一部の元素を無視するような
モデルを使って訓練データを生成する場合は、無視する元素を指定します。
vac_map 、 restart については現状対応していないので、
例のように設定してください。
aenet用の入力ファイル¶
aenet用の入力ファイルを [train] セクションの base_input_dir で
設定したディレクトリ内の generate 、 train 、および predict
ディレクトリに設置します。
generate¶
aenetでは、訓練用の原子配置とエネルギーのデータを、原子環境記述子とエネルギーの関係に変換した中間バイナリフォーマットにまとめてから訓練を行います。
この変換を行う generate.x 用の入力ファイルを generate ディレクトリに設置します。
まず、原子種ごとの記述子設定ファイルを用意します。ファイル名は任意ですが、チュートリアルでは
Al.fingerprint.stp , Mg.fingerprint.stp という名前にしています。
例として Al.fingerprint.stp の内容を示します:
DESCR
N. Artrith and A. Urban, Comput. Mater. Sci. 114 (2016) 135-150.
N. Artrith, A. Urban, and G. Ceder, Phys. Rev. B 96 (2017) 014112.
END DESCR
ATOM Al # 元素を指定
ENV 2 # ATOMで指定した元素と相互作用する原子種の数と元素名を指定
Al
Mg
RMIN 0.55d0 # 原子間の最隣接距離
BASIS type=Chebyshev # チェビシェフ記述子の設定
radial_Rc = 8.0 radial_N = 16 angular_Rc = 6.5 angular_N = 4
記述子設定の詳細についてはaenetのドキュメントを参照してください。
次に、
generate.in.head という名前で以下の内容のファイルを準備します:
OUTPUT aenet.train
TYPES
2
Al -0.0 ! eV
Mg -0.0 ! eV
SETUPS
Al Al.fingerprint.stp
Mg Mg.fingerprint.stp
OUTPUT には必ず aenet.train を指定してください。
TYPES 以下には訓練データ中の原子種とその数を指定します。
原子種ごとにエネルギーの基準を指定することもできますが、基本的には0に設定しておくのが無難です。
SETUPS 以下には原子種ごとの記述子設定ファイルを指定します。
ファイルの末尾には必ず改行が入っていることを確認してください。
abICSは generate.in.head の末尾に座標ファイルのリストを追加して generate.in を生成し、
generate.x を実行します。
train¶
generate で生成された訓練データを読み込み、訓練を行う
train.x 用の入力ファイルを train ディレクトリに設置します。
ファイル名は train.in としてください:
TRAININGSET aenet.train
TESTPERCENT 10
ITERATIONS 500
MAXENERGY 10000
TIMING
!SAVE_ENERGIES
METHOD
bfgs
NETWORKS
! atom network hidden
! types file-name layers nodes:activation
Al Al.15t-15t.nn 2 15:tanh 15:tanh
Mg Mg.15t-15t.nn 2 15:tanh 15:tanh
基本的には、 NETWORKS セクション以外は変更の必要はありません。
NETWORKS セクションでは、生成する原子種ごとのポテンシャル
ファイル名と、ニューラルネットワーク構造、および活性化関数を指定します。
predict¶
訓練したポテンシャルモデルを使って入力座標に対してエネルギーを
評価するための predict.x 用の入力ファイル predict.in を、 predict
ディレクトリに設置します:
TYPES
2
Mg
Al
NETWORKS
Mg Mg.15t-15t.nn
Al Al.15t-15t.nn
VERBOSITY low
TYPES セクションには原子種の数と元素名を、
NETWORKS セクションには原子種ごとのポテンシャルファイル名( train.in で設定したもの)を入力してください。
また、 VERBOSITY は必ず low に設定してください。
[sampling.solver]セクションのpathを実行環境における aenet のpredict.xのパスに設定する。[sampling.solver]と[train]セクションでignore_species = ["O"]を指定する。
計算実行¶
入力ファイルの準備・設定が完了後、実際に計算する方法について説明します。
サンプルスクリプトには、計算手順を簡略化するためのスクリプト AL.sh が準備されています。
run_pw.sh はQEの計算を実行するためのスクリプトで、後述する parallel_run.sh 内部で呼び出されます。
AL.sh の中身は以下の通りです。
#!/bin/sh
#SBATCH -p i8cpu
#SBATCH -N 4
#SBATCH -n 512
#SBATCH -J spinel
#SBATCH -c 1
#SBATCH --time=0:30:00
# Run reference DFT calc.
echo start AL sample
srun -n 8 abics_mlref input.toml >> abics_mlref.out
echo start parallel_run 1
sh parallel_run.sh
echo start AL final
srun -n 8 abics_mlref input.toml >> abics_mlref.out
#train
echo start training
abics_train input.toml >> abics_train.out
echo Done
先頭の #SBATCH で始まる数行は物性研スパコンでのジョブスケジューラに関するコマンドです。
ここでは、プロセス数512のMPI並列を指定しています。
また、 srun はスパコンの並列環境でプログラムを実行するためのコマンドです( mpiexec に相当します)。
ジョブスケジューラに関する詳細は、実際に利用する計算機システムのマニュアルを参照してください。
# Run reference DFT calc.
echo start AL sample
srun -n 8 abics_mlref input.toml >> abics_mlref.out
まず、 abics_mlref を用いて訓練データの大元となる第一原理計算用の入力ファイルを生成します。
初回実行時は、指定した数の原子配置をランダムに生成し、
それぞれの原子配置ごとに個別のディレクトリを用意した上で、入力ファイルをその中に作成します。
あわせて、これらのディレクトリのpathを記載したファイル rundirs.txt を生成します。
次に、得られたファイルをもとに第一原理計算を実行します。
echo start parallel_run 1
sh parallel_run.sh
parallel_run.sh は、GNU parallelを用いてQEの網羅計算を行うためのスクリプトで、
rundirs.txt に記載されたディレクトリを対象にQEの網羅計算が行われます。
QEの計算結果はそれぞれのディレクトリに格納されます。
QEの網羅計算により教師データが作成されましたので、次にaenetでのニューラルネットワークポテンシャルの作成に移ります。
最初に、 abics_mlref を再度実行し、第一原理計算の結果をabics_trainが読み込む共通フォーマットに変換したファイルを作成します。
echo start AL final
srun -n 8 abics_mlref input.toml >> abics_mlref.out
次に、学習データをもとにaenetによりニューラルネットワークポテンシャルの作成を行います。
ニューラルネットワークポテンシャルの計算には abics_train を使います。
[train] セクションの base_input_dir で指定したディレクトリに格納された入力ファイルを読み込み、計算を行います。
計算が無事終了すると、 baseinput ディレクトリに学習済みのニューラルネットワークが出力されます。
#train
echo start training
abics_train input.toml >> abics_train.out
以上の手続きで、能動学習を行うための AL.sh のプロセスが完了します。
4.2. モンテカルロサンプリング¶
次に、学習したニューラルネットワークポテンシャルを用い、abICSによりモンテカルロサンプリングをします。
4.2.1. 入力ファイルの準備¶
abICS でモンテカルロサンプリングを行うにはabICS制御ファイルでパラメータの設定をする必要があります。
abICS制御ファイル (input.toml)¶
レプリカ交換モンテカルロ法に関連するセクション [sampling] で計算パラメータを設定します。
(i) [sampling] セクション¶
[sampling]
nreplicas = 8
nprocs_per_replica = 1
kTstart = 600.0
kTend = 2000.0
nsteps = 6400
RXtrial_frequency = 4
sample_frequency = 16
print_frequency = 1
reload = false
レプリカ交換モンテカルロ(RXMC)法のレプリカの数や温度範囲などに関する設定を行います(入力ファイルフォーマット)。
今回は、RXMC計算のエネルギーソルバーとしてaenetの predict.x を用います。
現状、MPI版の predict.x はサポートしていないため、 nprocs_per_replica は
1を指定してください。
基本的に、 nreplicas は [mlref] セクションと同じ値にしてください。
(ii) [sampling.solver] セクション¶
[sampling.solver] # RXMC計算に使うソルバーの設定
type = 'aenet'
path= 'predict.x-2.0.4-ifort_serial'
base_input_dir = './baseinput'
perturb = 0.0
run_scheme = 'subprocess'
ignore_species = ["O"]
RXMC計算に使うエネルギーソルバーの設定を行います。今回は、aenetを使ってニューラルネットワークモデルの評価を行います。
type , perturb , run_scheme に関しては、能動学習スキームを用いる場合は上の例のまま変更しないでください。
path には、実行環境におけるaenetの predict.x のパスを指定してください。 base_input_dir
は自由に設定して構いません。
設定したディレクトリの中に predict.x に対応した入力ファイルが自動で設置されます(後述)。
ignore_species では、
ニューラルネットワークモデルで「無視」する原子種を指定できます。今回の例題では、Oの副格子は常に占有率1なので、Oの
配置はエネルギーに影響を及ぼしません。こういった場合は、ニューラルネットワークモデルの訓練および評価時に存在を無視した方が、
計算効率が高くなります。
4.2.2. 計算実行¶
サンプルスクリプトには、計算手順を簡略化するためのスクリプト MC.sh が準備されています。
MC.sh スクリプトの中身は以下の通りです。
#!/bin/sh
#SBATCH -p i8cpu
#SBATCH -N 1
#SBATCH -n 8
#SBATCH --time=00:30:00
srun -n 8 abics_sampling input.toml >> aenet.out
echo Done
abics_sampling を実行すると MCxx ディレクトリが作成されます (xxは実行回数)。
abICS では、active learning 向けに、ALloop.progress ファイルから計算回数などの情報を取得する機能が追加されています。
MCxx ディレクトリの下には、レプリカ数分だけのフォルダが作成され、
VASPのPOSCARファイル形式で記載された各ステップごとの原子配置(structure.XXX.vasp)、
最低エネルギーを与えた原子位置(minE.vasp)や、
各ステップごとの温度とエネルギー(obs.dat)などが出力されます。
詳細については abICSマニュアルの出力ファイル を参考にしてください。
上の手続きで得られた結果は、aenetにより求められたニューラルネットワークポテンシャルの精度に依存します。 はじめのステップではランダムな配置をもとに学習を行ったので、低温の構造については精度が低いことが予想されます。 そこで、モンテカルロで推定された構造に対して、再度第一原理計算でエネルギーを計算し再学習させます。 このステップを繰り返すことで、全温度領域での精度を高めることが期待されます。
このプロセスは、 AL.sh と MC.sh を順番に繰り返すことで計算できます。
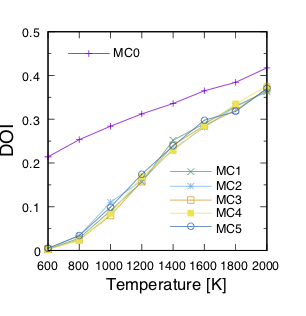
実際に下図に反転率(DOI)を計算した結果を掲載します。
この例では、最初の一回目の結果がMC0、その後MC1, MC2, ..., MC5と5回実行させています。
最初の一回目が他のものとかなりずれていることから、精度が出ていないことが見てとれます。
一度モンテカルロを行った結果を元に学習させると、その次からはほぼ同じような値が得られていることがわかります。
なお、DOIについては以下の手順で計算できます。
MCxx ディレクトリに移動する。
srun -n 8 abicsRXsepT ../input.tomlを実行してTseparateディレクトリを作成する (並列数はabics_samplingを実行した際の並列数に揃える。本チュートリアルでは並列数を8にしているので8に設定)。sampleディレクトリにある
calc_DOI.pyとMgAl2O4.vaspをコピーする。
srun -n 8 python3 calc_DOI.py ../input.tomlを実行して温度ごとの反転度を計算する。 (並列数の指定は 2. と同様)。
一般的には、MCxxx/Tseparate``の中に入っている温度ごとの構造ファイルから所望の熱力学平均を計算するスクリプト
(今回の場合は``calc_DOI.py)をユーザーが用意する必要があります。
また、反転度の計算を完全に収束させるためには、例題のモンテカルロステップ数では不十分であることにご注意ください。 能動学習のサイクルとは別にモンテカルロステップ数を増やした計算を行って、熱力学平均を計算することをおすすめ します。
4.2.3. LAMMPS インターフェースを利用したサンプリング¶
モンテカルロサンプリングにおいては、 LAMMPSインターフェースをもちいた aenet ライブラリ呼び出しにも対応しています(aenetPyLammps ソルバー)。
ファイル入出力などを行わないため、 aenet をプロセスとして呼び出すよりも高速に動作します。
入力ファイル例としては examples/active_learning_qe_lammps を参考にしてください。
aenet-lammps のインストール¶
aenetPyLammps の利用には、 aenet-lammps を組み込んだ LAMMPS のインストールが必要です。
コミット 5d0f4bc で動作確認済。
git checkout 5d0f4bc
上記URLで指定された手順に従ってインストールしてください。以下、インストール時の注意事項です。
aenetmakefiles/Makefile.*中のFCFLAGSオプションに-fPICを追加してください。
lammpssrc/Makefile中にLMP_INC = -DLAMMPS_EXCEPTIONSを追加してください。make 時にオプションで
mode=sharedをつけるようにしてください。
上記のインストール終了後、
make install-pythonを実行してください。pythonコマンドで起動するPython 環境にlammpsパッケージがインストールされます。
モデル学習¶
モデル学習のやりかたは前述の aenet によるものと同様です。
サンプリング¶
predict 用入力ファイル¶
predict.x でつかわれていた入力ファイル predict.in のかわりに、以下のような入力ファイル in.lammps を predict ディレクトリに設置してください:
pair_style aenet
pair_coeff * * v00 Al Mg 15t-15t.nn Al Mg
neighbor 0.1 bin
入力ファイルフォーマットの詳細は aenet-lammps リポジトリの README を参照してください。
サンプリング¶
[sanmping.solver] セクションの type を 'aenetPyLammps' に、 run_scheme を 'function' に設定すると、 LAMMPS インターフェースを利用したサンプリングを実行できます。
[sampling.solver]
type = 'aenetPyLammps'
base_input_dir = ['./baseinput']
perturb = 0.0
run_scheme = 'function'